
Scleroza laterală amiotrofică este o afecţiune neurodegenerativă multifactorială, care necesită o abordare terapeutică complexă, bazată în primul rând pe îmbunătăţirea calităţii vieţii pacienţilor.
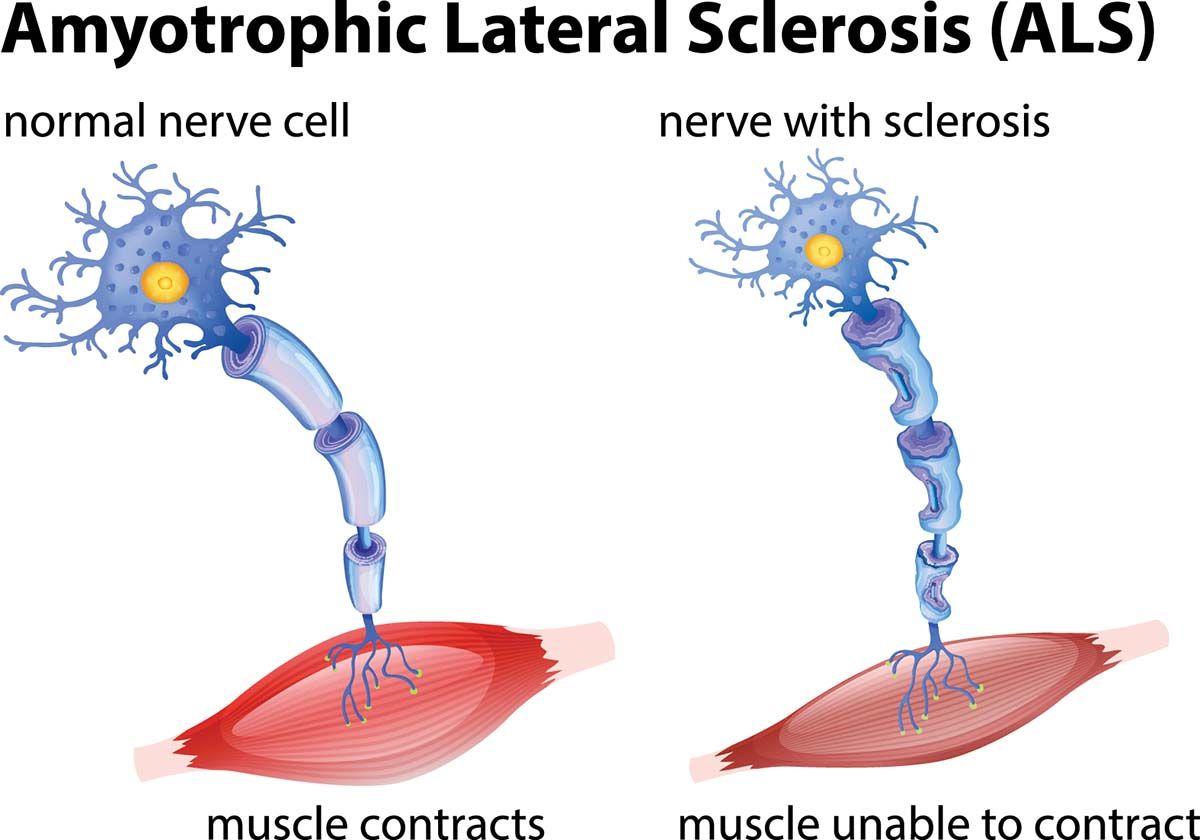
Scleroza laterală amiotrofică (SLA) sau boala neuronului motor (BNM), cunoscută și ca boala lui Lou Gehrig, este o afecţiune neurodegenerativă progresivă, fatală în 1-5 ani de la debut, caracterizată prin degenerarea neuronilor motori centrali și periferici, determinând simptome motorii și non-motorii (1-3).
Afectează persoanele de orice rasă/etnie, dar în populaţia europeană este mai frecventă la bărbaţi decât la femei (1,5/1). Persoanele cu vârste cuprinse între 50 și 75 de ani și cei cu istoric familial de SLA au un risc mai mare de a dezvolta boala (2,4).
SLA are două forme de prezentare: o formă sporadică, cel mai frecvent întâlnită (95%), și una familială, care însumează doar 5-10% din totalul cazurilor, dintre care 20% au transmitere autozomal dominantă (4-6).
De cele mai multe ori, etiologia sclerozei laterale amiotrofice nu este cunoscută, dar există cazuri de boală familială asociate cu mutaţii la nivelul unor gene precum SOD1, FUS/TLS, TARDBP 43 și C9orf72. SLA poate fi considerată o boală multifactorială în care un rol important îl are interacţiunea dintre factorii genetici și factorii externi (1,5). Dintre factorii de mediu evaluaţi, amintim expunerea la metale grele, câmpuri electromagnetice, la pesticide, activitatea fizică intensă și fumatul (4,5).
Cele mai importante caracteristici patologice evidenţiate în SLA sunt reprezentate de atrofia mușchilor scheletici, atrofia cortexului motor, paloarea și scleroza tracturilor piramidale (tract corticospinal, tract corticobulbar), dar și subţierea atât a nervilor hipogloși, implicaţi în controlul musculaturii limbii, cât și a rădăcinilor ventrale ale măduvei spinării. Studiile microscopice arată o depleţie a peste 50% dintre neuronii motori spinali și o glioză astrocitară difuză și infiltrare microglială în substanţa albă și cenușie a măduvei (1).
profesor la catedra de matematică de la Universitatea Cambridge, autor de volume
de popularizare a fizicii) a fost diagnosticat cu SLA la vârsta de 21 de ani. A trăit 76 de ani.
Fenotipul bolii este clasificat în funcţie de localizarea simptomelor de debut. Astfel, cei mai mulţi pacienţi (65%) se prezintă cu simptome debutate la nivelul membrelor (forma spinală), în timp ce 30% prezintă semne de disfuncţie bulbară, precum disfagie și dizartrie, iar 5% dintre pacienţi au debut respirator.
În general, debutul simptomatologiei este asimetric, iar examinarea neurologică evidenţiază o asociere a semnelor de NMC (neuron motor central) și NMP (neuron motor periferic), cele mai frecvente manifestări clinice fiind slăbiciunea musculară, crampele musculare, fasciculaţii, hipertonia spastică și hiperreflexia (4,5).
Caracteristic pentru SLA este că motilitatea extraoculară și controlul sfincterian sunt cruţate, cel puţin până în stadiile avansate ale bolii, iar la examinarea sensibilităţii s-au descris rar semne minore. O altă caracteristică frecvent întâlnită în rândul pacienţilor cu SLA este deteriorarea cognitivă – demenţa frontotemporală, care apare la aproximativ 15% dintre pacienţi, fiind precedată de tulburări de
personalitate și iritabilitate (4).
În încercarea de a explica mecanismele care pot provoca sau contribui la dezvoltarea bolii, au fost propuse patru ipoteze primare: stresul oxidativ, idee susţinută de faptul că mutaţia la nivelul SOD1 este o cauză primară a SLA; strangularea axonală prin dezorganizare neurofilamentoasă, idee susţinută de acumularea anormală de neurofilamente ca o caracteristică patologică a multor cazuri de SLA sporadică sau familială mediată SOD1; toxicitatea agregatelor intracelulare și/sau eșecul degradării proteinelor, o caracteristică comună a SLA cu mutaţie la nivelul SOD1; și excitotoxicitatea, neuronii motori fiind foarte sensibili la toxicitatea indusă de influxul de Ca2+ cauzat de stimularea glutamatergică excesivă.
Indiferent de mecanismele care stau la baza dezvoltării SLA, rezultatul final este retracţia axonală și denervarea celulei ţintă. În cazul neuronului motor periferic (NMP), retracţia axonală determină denervarea mușchiului, iar în cazul neuronului motor central (NMC), retracţia axonală determină pierderea controlului asupra NMP, cu hipotonie și slăbiciune musculară (3,4).
Pacient în vârstă de 45 de ani, nefumător, se prezintă pentru un traumatism cranio-cerebral prin cădere de la înălţime, soldat cu pierderea stării de conștienţă și amnezie retrogradă și anterogradă în privinţa evenimentelor din ziua respectivă, motiv pentru care se internează în secţia noastră.
Din istoricul pacientului reţinem că simptomatologia a debutat în august-septembrie 2018 cu dorsolombalgie, urmată de crampe musculare crural de partea stângă. În luna noiembrie 2019 prezintă un singur episod de cădere de la același nivel cu fractură claviculară stângă secundară, iar începând din luna februarie 2020 – slăbiciune musculară la nivelul membrelor pelvine și instalarea tulburărilor de mers cu agravare progresivă.
Din examenul obiectiv neurologic la internare reţinem prezenţa fasciculaţiilor linguale, reflexe faringiene vii, frust deficit motor la nivelul membrului inferior stâng distal (4/5 MRC – scala Medical Research Council), fasciculaţii spontane și la stimulare la nivelul membrelor superioare și inferioare bilateral, mers posibil cu aspect de pareză pseudo-SPE (sciatic popliteu extern) – pacientul atinge podeaua cu partea antero-laterală a plantei stângi –, ROT vii global, asimetrice, RCP (reflex cutanat plantar) în flexie bilateral.
Istoric
Anterior internării în secţia noastră, pacientul a efectuat mai multe investigaţii imagistice în ambulatoriu ale căror rezultate însă nu explică prezenţa și progresia simptomatologiei:
Rezultatele testelor de laborator au fost în limite normale, cu excepţia unei ușoare leucocitoze (12,73 x 1.000/uL). Coroborând datele clinice și paraclinice, evaluările imagistice și studiile electrofiziologice, s-a luat în considerare diagnosticul de boala neuronului motor și s-a recomandat iniţierea tratamentului cu riluzole cp 50 mg x 2/zi, cu monitorizarea periodică a hemoleucogramei, ionogramei, funcţiei hepatice și renale.
În funcţie de distribuţia predominantă a simptomelor de debut (musculatură bulbară sau a membrelor) și în funcţie de tipul de neuroni motori afectaţi predominant (NMC, NMP), boala neuronului motor poate fi împărţită în cinci tipuri (6):
În plus, există mai multe modele de implicare neuromusculară. De exemplu, un picior poate fi afectat înaintea mâinii, astfel că un semn de „picior căzut“ (drop foot) însoţit de slăbiciune musculară și de atrofia mușchilor pretibiali poate fi incorect atribuit compresiei de nerv peronier până la apariţia slăbiciunii mușchilor gastrocnemieni și a altor mușchi, care evidenţiază afectarea mai extinsă a neuronilor lombosacraţi.
Un alt mod de prezentare este cel care implică afectarea precoce a musculaturii toracale, abdominale și musculaturii posterioare a gâtului, determinând căderea capului și camptocormia (n.r.: sindrom caracterizat prin încovoierea spatelui ca urmare a unei contracturi musculare). Mai este cunoscut un tip de afectare, care implică amiotrofia proximală și simetrică a membrelor și a centurilor, cu debut la vârste tinere, care simulează distrofia musculară.
Un alt mod de prezentare este prin afectarea precoce a musculaturii respiratorii (mușchi intercostali, diafragm), cu evoluţie rapidă spre insuficienţă respiratorie (7). Ca și în cazul prezentat, prima și cea mai evidentă manifestare a bolii poate fi slăbiciunea musculară la nivelul membrelor inferioare.
Astfel, în urma relatărilor pacientului cu privire la debutul simptomatologiei, putem obiectiva forma cu debut pseudo-SPE, cu slăbiciune musculară și atrofia mușchilor pretibiali.
Pe baza criteriilor El Escorial (tabel), putem include pacientul în forma definită de SLA, acesta prezentând atât semne de afectare a NMP din trunchiul cerebral (fasciculaţii linguale), semne de afectare a NMC din tractul corticobulbar (reflexe faringiene vii), cât și semne de afectare spinală în trei regiuni.

Printre investigaţiile paraclinice utile în boala neuronului motor se numără studiile de electroneuromiografie, care au evidenţiat descărcări spontane de denervare (potenţiale de fibrilaţie), fasciculaţii și unităţi polifazice, demonstrând astfel denervarea și reinervarea în teritoriile studiate, iar studiile de conducere nervoasă motorie au arătat doar o încetinire ușoară, fără bloc de conducere focal. Testele de conducere nervoasă motorie au viteze normale, iar amplitudinile de conducere devin progresiv mai mici pe măsură ce boala avansează (4,7).
Testele de laborator de rutină la un pacient cu SLA ar trebui să includă dozarea VSH, electroforeza proteinelor serice și urinare, evaluarea funcţiei tiroidiene, dozarea calciului seric și fosfatului, screening pentru metale grele, analiza LCR. În ceea ce privește cazul prezentat, rezultatele testelor de laborator au fost în limite normale, cu excepţia analizei LCR, care nu a fost realizată (4).
Citiți și: Managementul durerii în bolile reumatismale
Deși au fost studiate peste 50 de medicamente cu diferite mecanisme de acţiune pentru tratamentul SLA, doar două molecule (riluzole și edaravone) au ajuns pe piaţă. Riluzole a fost primul tratament aprobat pentru SLA, care acţionează prin reducerea neurotransmisiei glutamatergice prin blocarea canalelor de Na voltaj dependente la nivelul neuronilor presinaptici.
Administrarea orală a 100 mg riluzole pe zi prelungește supravieţuirea cu 3 luni după 18 luni de tratament. Edaravone se presupune că acţionează ca antioxidant și poate încetini progresia bolii, la pacienţi atent selecţionaţi, cu debut precoce și boală rapid progresivă (1,8).
Un aspect deosebit de important în managementul bolii neuronului motor este îmbunătăţirea calităţii vieţii pacienţilor prin iniţierea tratamentului simptomatic pe măsură ce simptomele devin evidente și invalidante (8,9).
Astfel, putem trata:
Preluare din volumul ,,Sistem nervos – Cazuri clinice interdisciplinare”, coordonat de prof. dr. Carmen Adella Sîrbu și ș.l. dr. Marian Mitrică, apărut la Editura Universitară „Carol Davila”, București, în 2020.
Bibliografie
1. Hardiman O, Al Chalabi A, Chio A, et al. Amyotrophic lateral sclerosis [published correction appears in Nat Rev Dis Primers. 2017 Oct 20;3:17085]. Nat Rev Dis Primers. 2017;3:17071. Published 2017 Oct 5
2. Mehta P, Kaye W, Raymond J, et al. Prevalence of Amyotrophic Lateral Sclerosis – United States, 2015. MMWR Morb Mortal Wkly Rep. 2018;67(46):1285 1289. Published 2018 Nov 23
3. Cleveland DW, Rothstein JD. From Charcot to Lou Gehrig: deciphering selective motor neuron death in ALS. Nat Rev Neurosci. 2001;2(11):806 819
4. Goutman SA. Diagnosis and Clinical Management of Amyotrophic Lateral Sclerosis and Other Motor Neuron Disorders. Continuum (Minneap Minn). 2017;23(5, Peripheral Nerve and Motor Neuron Disorders):1332 1359
5. Bozzoni V, Pansarasa O, Diamanti L, Nosari G, Cereda C, Ceroni M. Amyotrophic lateral sclerosis and environmental factors. Funct Neurol. 2016;31(1):7 19
6. Aminoff MJ, Greenberg DA, Simon RP. Clinical Neurology 9th edition, 2015, Lange, 245 248
7. Ropper AH, Samuels MA, Klein JP, Prasad S. Adams and Victor’s Principles of Neurology, 11th edition. 2019, McGraw Hill Education, 1133 1140
8. Băjenaru O. Ghiduri de diagnostic și tratament în neurologie, 2010, Amaltea, 239 263
9. Goutman SA, Simmons Z. Symptom management in amyotrophic lateral sclerosis: We can do better. Muscle Nerve. 2018;57(1):1 3
Dacă vrei să fii la curent cu tot ce se întâmplă în lumea medicală, abonează-te la „Viața Medicală”, publicația profesională, socială și culturală a profesioniștilor în Sănătate din România!
Titularii abonamentelor pe 12 luni sunt creditați astfel de:

Dacă vrei să fii la curent cu tot ce se întâmplă în lumea medicală, abonează-te la „Viața Medicală”, publicația profesională, socială și culturală a profesioniștilor în Sănătate din România!
Află mai multe informații despre oferta de abonare.
Cookie-urile ne ajută să vă îmbunătățim experiența pe site-ul nostru. Prin continuarea navigării pe site-ul www.viata-medicala.ro, veți accepta implicit folosirea de cookie-uri pe parcursul vizitei dumneavoastră.
Da, sunt de acord Aflați mai multe








.jpg)





