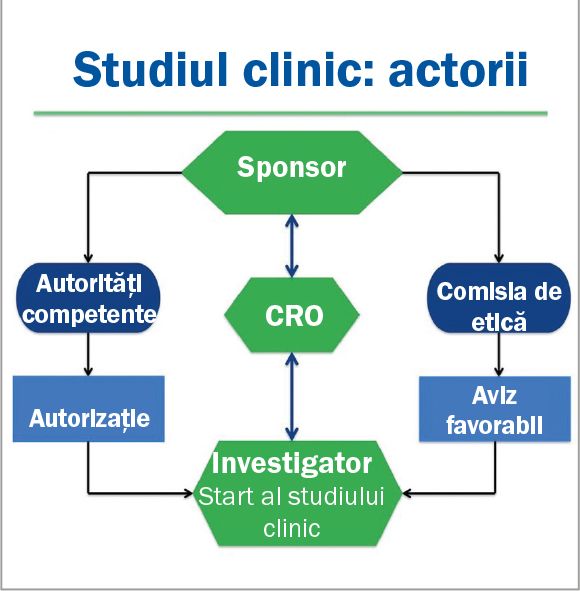
Orice studiu clinic presupune lucrul într-o echipă formată din sponsorul farma sau biotech, investigator, monitor, laborator, autorităţile de reglementare etc. Responsabilităţile acestora sunt foarte bine definite în GCP (Good Clinical Practice, Ghidul de bună practică clinică), după care se conduc toate proiectele de cercetare clinică. Astăzi vorbim despre rolurile sponsorului farma.
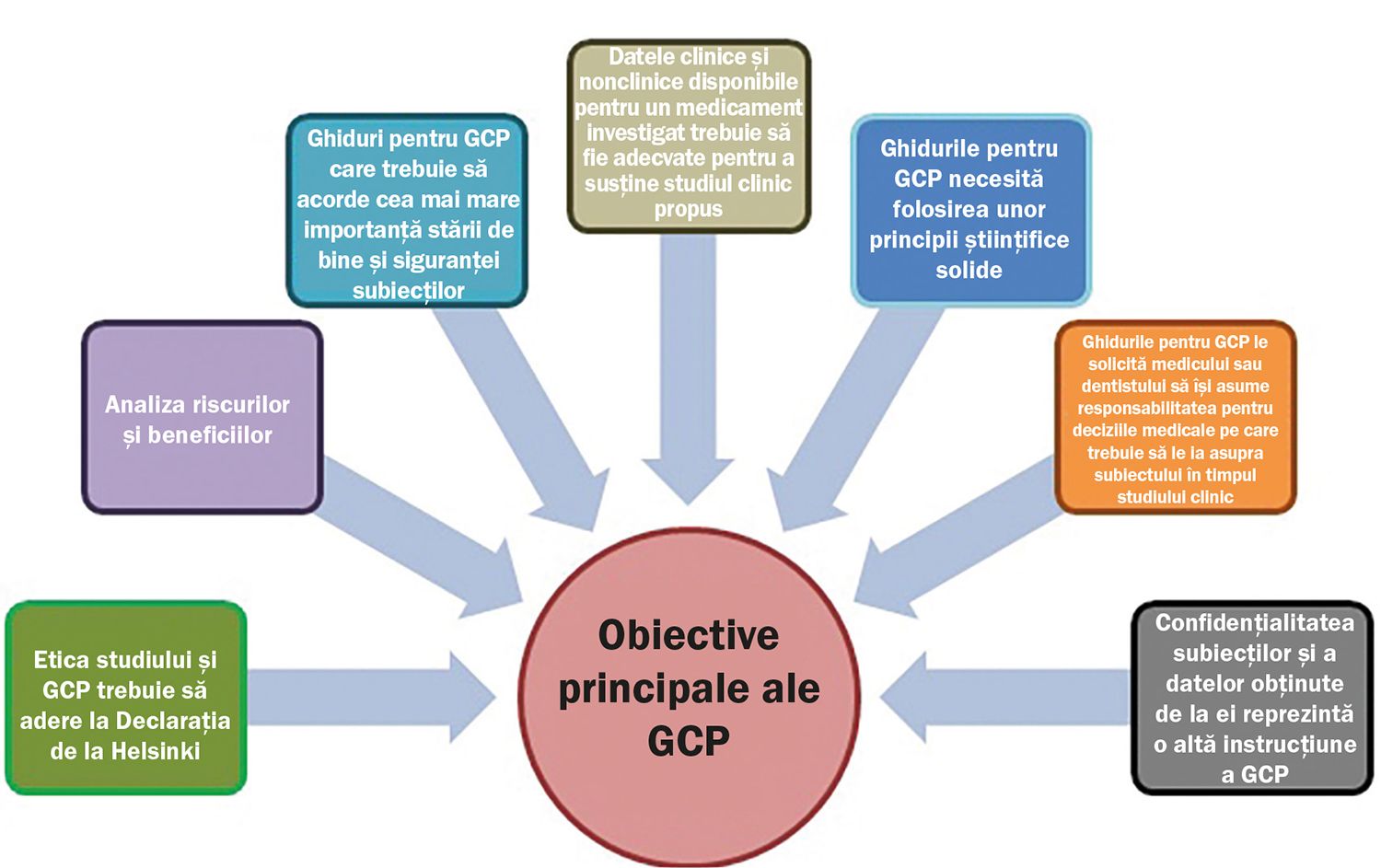
După ce lumea medicală a fost zguduită de evenimente tragice, cu consecinţe grave asupra pacienţilor, cu pierderi importante de vieţi, s-a ajuns la concluzia că toate cercetările clinice pe subiecţi umani trebuie să fie foarte atent observate și controlate, desfășurându-se conform codului GCP. Cum s-a ajuns aici? Să trecem prin evenimentele marcante.
În 1937 are loc una din cele mai mari otrăviri în masă din secolul XX (1), apărută după introducerea sulfanilamidei, primul medicament antimicrobian. Diluantul folosit în preparare, dietilen glycol, ucide 106 pacienţi, o treime fiind copii (n.r.: The Elixir Tragedy).
În 25 iunie 1938, agenţia FDA (Food and Drug Agency, USA), operaţională ca agenţie de reglementare încă din 1930, emite documentul „Federal Food, Drug and Cosmetic Act” (2) care o autorizează să efectueze inspecţii la nivelul producătorilor de medicamente. După experimentele monstruoase efectuate în lagărele de concentrare din Germania în timpul celui de-al Doilea Război Mondial, „The Nuremberg Code (from Trials of War Criminals before the Nuremberg Military Tribunals under Control Council Law No. 10, October 1946-April 1949, Washington, D.C)” explică modul în care sunt permise experimentele realizate pe subiecţi umani. Drept urmare, în 1948 apare Declaraţia Universală a Drepturilor Omului, iar în 1950 are loc The European Convention for the Protection of Human Rights and Fundamental Freedoms.
O altă tragedie produce Thalidomida (3), medicament cu efect sedativ folosit și în tratamentul mielomului multiplu. Între anii 1950 și 1960, peste 10.000 de copii din întreaga lume se nasc cu malformaţii congenitale (fără membre, cu coloana vertebrală contorsionată, cu surditate sau cecitate), după ce mamele lor au folosit Thalidomida ca antiemetic împotriva stărilor de greaţă din primul trimestru de sarcină. Are loc cel mai mare scandal internaţional din industria farmaceutică de după cel de-al Doilea Război Mondial. Ca urmare, în 1962, în SUA apare Amendamentul Kefauver-Harris, care solicită trimiterea către FDA și investigarea amănunţită a produsului de investigat (IP/investigational product) de către agenţie, înainte de orice fel de campanie de marketing și a scoaterii pe piaţă pentru publicul larg.
În 1964, Declaraţia de la Helsinki creată de World Medical Association arăta că scopul cercetării medicale care implică subiecţi umani este acela de a îmbunătăţi diagnosticul, terapia și măsurile profilactice, printr-o înţelegere mai bună a etiologiei și patogeniei bolilor.
În 1996, ca urmare a unei iniţiative comune a FDA, EMA (Agenţia Europeană a Medicamentului) și a Ministerului Sănătăţii din Japonia, apare ghidul unic de bune practici pentru cercetarea clinică – ICH-GCP (International Conference of Harmonisation – Good Clinical Practice). Publicat pe 16 ianuarie 1997, acesta definește foarte clar toate cerinţele tehnice necesare înregistrării medicamentelor de uz uman. Ghidul ICH-GCP este un standard în care se explică foarte clar cum studiile clinice sunt create cu un anume design și cum trebuie implementate și raportate către autorităţi. GCP ghidează și explică responsabilităţile sponsorilor, monitorilor, investigatorilor, comisiilor de etică sau autorităţilor competente din fiecare ţară unde se desfășoară studiul clinic.
Sponsorul (compania farmaceutică) are întreaga responsabilitate pentru iniţierea, managementul și finanţarea studiului clinic. Compania farmaceutică este responsabilă cu:
Sponsorul trebuie să:
Sponsorii nu pot face toate aceste lucruri singuri și de multe ori apelează la companii conducătoare de studii CRO/companii de monitorizare, care acţionează în numele lor și se asigură că drepturile și starea de bine a subiecţilor înrolaţi sunt protejate, datele raportate sunt complete și curate, iar acestea pot fi verificate oricând cu datele din documentele-sursă.
Azi, lumea medicală așteaptă cu ardoare să fie aprobate un nou medicament și un vaccin împotriva infecţiei cu virusul SARS-CoV-2 care cauzează COVID-19. În martie 2020, FDA a emis un ghid (4) de conducere a studiilor clinice în timpul pandemiei („FDA Guidance on Conduct of Clinical Trials of Medical Products during COVID-19 Pandemic Guidance for Industry, Investigators, and Institutional Review Boards - March 2020”) pentru „a da recomandări, iar sponsorii să susţină companii farmaceutice în asigurarea siguranţei subiecţilor studiilor lor, în acord cu GCP, și pentru a minimiza riscul privind integritatea studiului în timpul pandemiei
COVID-19”.
Ghidul oferă indicaţii despre modul de continuare a studiilor clinice aflate în desfășurare și se asigură că siguranţa pacienţilor rămâne principalul obiectiv al cercetării clinice în timpul pandemiei. FDA recomandă strânsa cooperare cu comisiile de etică privind toate modificările necesare de implementat ca urmare a COVID-19, în special în legătură cu eficacitatea medicamentului de studiat, explicarea necesităţii de a face modificări în protocol și ce impact are acest lucru asupra pacienţilor înrolaţi. Procedurile uzuale de monitorizare, incluzând vizitele de monitorizare la centrele de investigaţie, nu vor mai fi posibile, de aceea sponsorii farma trebuie să găsească soluţii alternative, ca vizitele efectuate virtual, utilizarea laboratoarelor și centrelor de imagistică locale, mai mult decât cele centrale, să ofere vizite la domiciliul pacienţilor, dacă este posibil, cu efectuarea de recoltări, precum și alte proceduri de studiu care anterior erau făcute la sediu și care acum nu mai sunt posibile. Totul pentru a menţine studiul activ, fără a crea discontinuităţi.
Un ghid similar a fost emis în 20 martie 2020 și de EMA (5) („Guidance to sponsors on how to manage clinical trials during the COVID-19 pandemic”). Acesta a fost agreat între CTEG (Clinical Trials Experts Group), CTFG (Clinical Trials Facilitation and Coordination Group) și GCP Inspectors’ Working Group și vine cu o abordare armonizată a modului în care să se desfășoare studiile clinice în această perioadă, pentru a reduce efectul negativ al pandemiei asupra bunului mers al acestora.
Dacă vrei să fii la curent cu tot ce se întâmplă în lumea medicală, abonează-te la „Viața Medicală”, publicația profesională, socială și culturală a profesioniștilor în Sănătate din România!
Titularii abonamentelor pe 12 luni sunt creditați astfel de:

Dacă vrei să fii la curent cu tot ce se întâmplă în lumea medicală, abonează-te la „Viața Medicală”, publicația profesională, socială și culturală a profesioniștilor în Sănătate din România!
Află mai multe informații despre oferta de abonare.
Cookie-urile ne ajută să vă îmbunătățim experiența pe site-ul nostru. Prin continuarea navigării pe site-ul www.viata-medicala.ro, veți accepta implicit folosirea de cookie-uri pe parcursul vizitei dumneavoastră.
Da, sunt de acord Aflați mai multe




.jpeg)