Rahitismele genetice sunt maladii generate de tulburări ale metabolismului fosfocalcic.
Concentraţia sărurilor fosfocalcice este influenţată de mai mulţi factori care depind de glandele endocrine, de tubul digestiv, ficat și rinichi. Fiziopatologia acestora poate genera tulburări funcţionale, ca urmare a unor boli genetice care interesează aceste aparate și organe. Rahitismele genetice sunt forme de rahitism rezistente în grad variabil la tratamentul cu vitamina D, motiv pentru care se mai numesc și rahitisme vitamino-D-rezistente. Studiile genetice au permis individualizarea mai multor forme de rahitism, unele dintre ele suprapunându-se peste tipuri de rahitism descrise anterior, eponimic sau ca idiopatice.
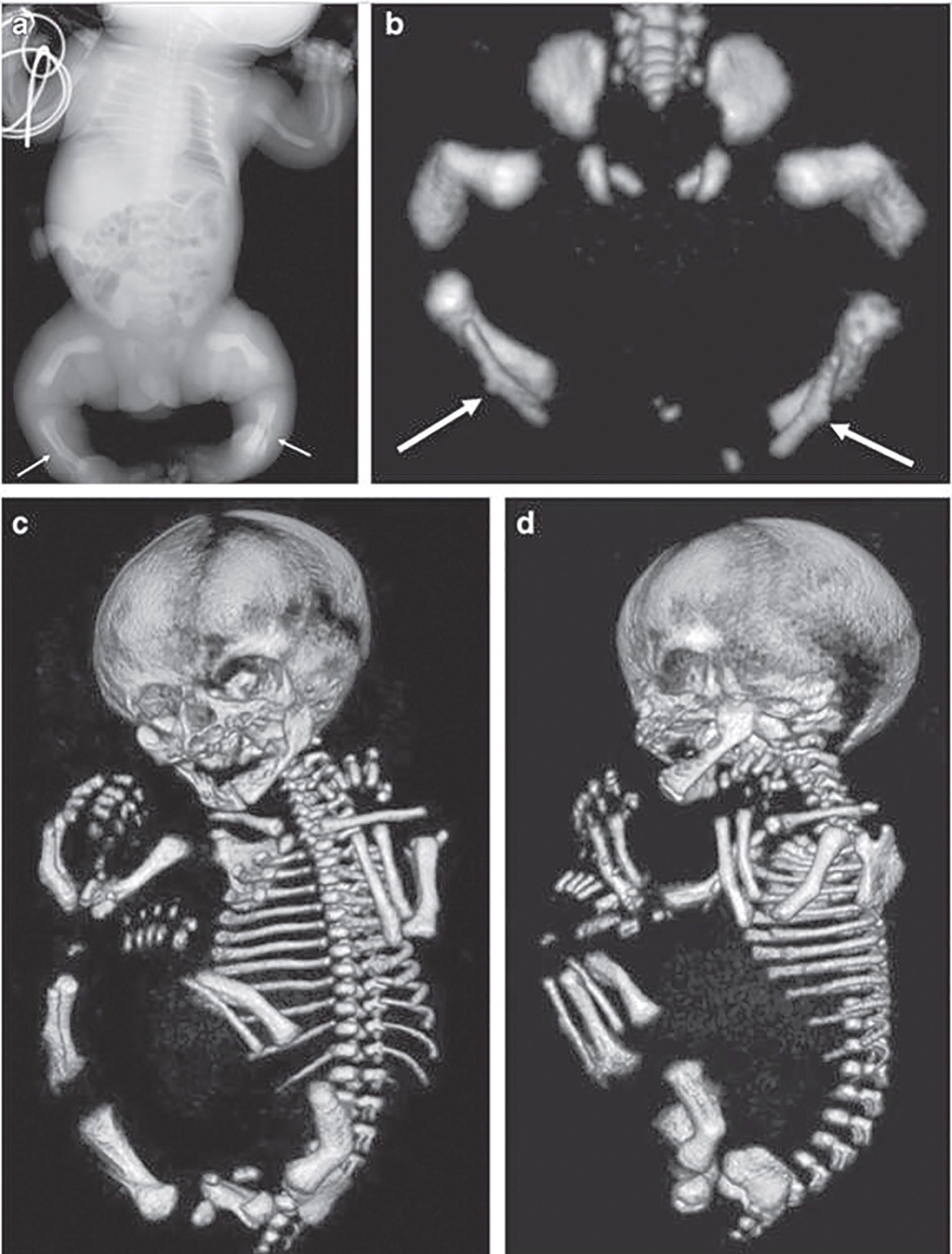
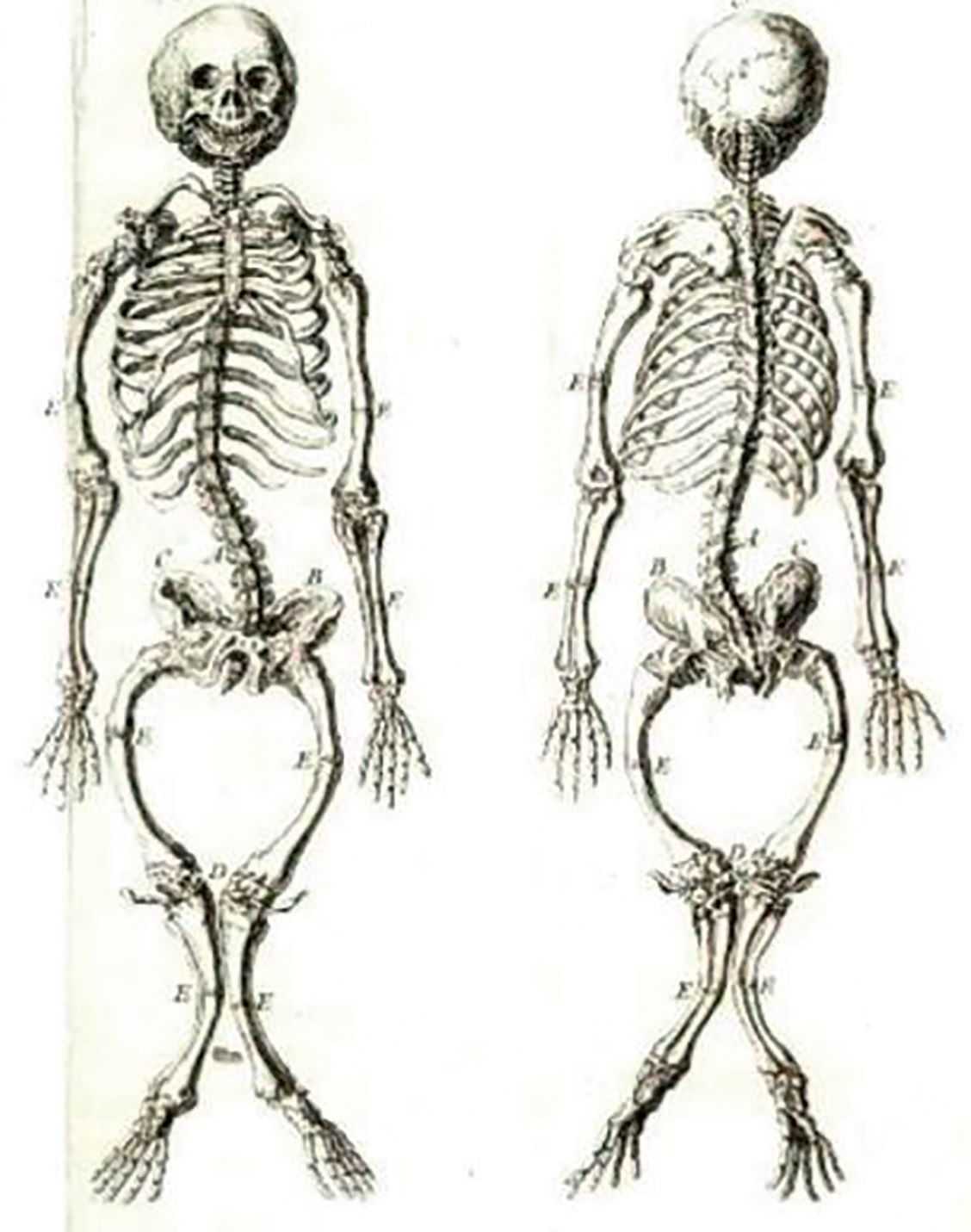
Patologia metabolismului fosfocalcic a suscitat interesul medicilor din cele mai vechi timpuri, din cauza efectelor sale secundare, care au stârnit curiozitatea în mediul social. S-au păstrat multe schelete din acea vreme, iar astăzi ele reprezintă prototipul evoluţiei spontane, fără tratament, a afecţiunii (fig. 1).
Studiile de fiziologie și fiziopatologie ale metabolismului fosfocalcic au contribuit la obţinerea de ameliorări în tratamentul rahitismului, mai ales în rahitismul carenţial. Expunerea la soare a dus la diminuarea cazurilor de osteomalacie. În ultimii ani au fost selectate cazurile de rahitism vitamino-D-rezistent, iar recent, studiile genetice au arătat că aceste forme sunt expresia unor mutaţii genetice. Astfel s-au perfectat metodele de diagnostic molecular, care vor permite în viitor tratamentul prin implanturi genetice. Studiile genetice permit selectarea cazurilor de rahitism genetic și diagnosticul formelor severe și letale. În acest domeniu și-au adus contribuţia S. Acar, S. Mumm, P. Glanc, Y. Shi, C.M. Rumack, J. Jones, S. Unger, K. Demir, Rathbun, Marotoux și alţii.
La noi în ţară strălucește de la distanţă activitatea profesorului N. Suciu, care prin activitatea în laboratoarele Centrului de Diagnostic Molecular contribuie la diagnosticul perinatal al afecţiunilor genetice și stabilește conduita practică pentru îngrijirea mamei și a copilului. În mod similar își aduc contribuţia prof. dr. Gheorghe Peltecu, prof. dr. N. Cernea și prof. dr. R. Vlădăreanu.
Rahitismele genetice sunt afecţiuni studiate nu numai de geneticieni și obstetricieni-ginecologi, ci și de pediatri, ortopezi pediatri, ortopezi și traumatologi, medici interniști, reumatologi, radiologi-imagiști etc.
În domeniul ortopediei pediatrice, prof. dr. A. Pesamosca și P. Moroz și-au adus o contribuţie remarcabilă în tratamentul copiilor cu rahitism genetic și cu manifestări scheletice. P. Moroz are cea mai mare cazuistică de copii cu rahitism genetic. A. Pesamosca prefera osteotomiile indinţate, tip Dobosiu sau Romke, care îi permiteau corecţia fără a folosi materiale de osteosinteză. El a instituit ca indicaţie operatorie în clinică tratamentul chirurgical numai după vârsta de 7 ani. Nimeni nu mai avea voie să intervină operator până la această vârstă. Astfel s-a reușit selectarea cazurilor vitamino-D-rezistente care la acea vreme nu erau cunoscute ca fiind genetice și s-a redus semnificativ numărul de intervenţii chirurgicale pentru această boală. Această indicaţie a fost preluată și de celelalte clinici din ţară și se păstrează și astăzi.
Alături de el, și-au adus contribuţia alţi profesori și medici de renume, cu o vastă experienţă în domeniu: M. Socolescu, A. Mironescu, A. Varna, Z. Moldovan, D. Tica, D. Goţia, G. Aprodu, P. Ţepeneu, I. Ionescu etc.
Profesorul Pesamosca a început alungirea membrelor la pacienţii cu nanism rahitic. Iniţial a alungit gambele cu 5 cm la un nanic hipofosfatemic la care a corectat simultan și încurbarea tibiei. Mai târziu s-au făcut alungirile la mai mulţi nanici cu rahitism: la o pacientă s-a obţinut alungirea cu 25 cm pe fiecare membru pelvin. Toţi acești pacienţi, alături de alte cazuri, au intrat în cazuistica lucrării mele de doctorat.
Rahitismele genetice X-linkate sunt determinate de inactivarea genei PHEX din cromozomul X. Această mutaţie produce dezechilibru în metabolismul fosfocalcic prin pierderea fosfaţilor prin urină, care se asociază cu hipofosfatemie, semn caracteristic în acest tip de rahitisme. Au fost detectate trei mutaţii genetice responsabile de rahitismul hipofosfatemic X-linkat: C59S, Q394X și W602. Examenul molecular poate evidenţia deleţia braţelor lungi ale cromozomului și pierderea unor exoni care pot genera diverse tipuri de rahitism, cu manifestări polimorfice.
Rahitismul hipofosfatemic X-linkat este o afecţiune genetică, vitamino-D-rezistentă, care are caracteristic nivelul scăzut al fosforului în sânge. Este o formă dominantă X-linkată de rahitism, diferită de celelalte forme prin rezistenţa la tratamentul cu vitamina D chiar la doze mari sau excesive. Nivelul scăzut al fosfatemiei se produce din cauza pierderii fosforului în urină, care determină tulburări în metabolismul fosfocalcic urmate de scăderea rezistenţei osoase și de apariţia deviaţiilor axiale ale membrelor.
Această afecţiune este determinată de mutaţia genei PHEX, cu localizare Xp22.2-p22.1 în cromozomul X. Se transmite autosomal dominant, diferenţiat în funcţie de părintele afectat.
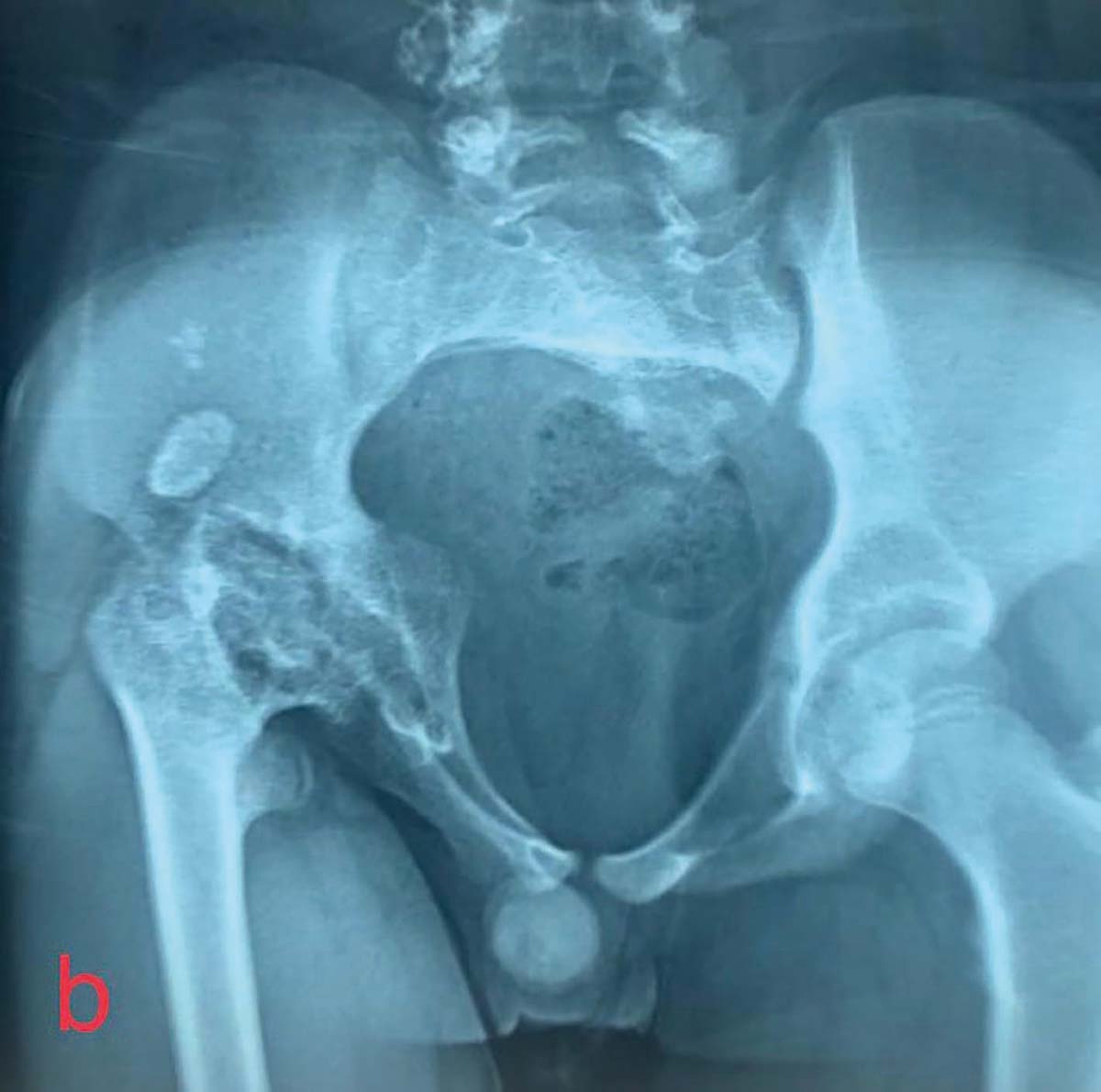
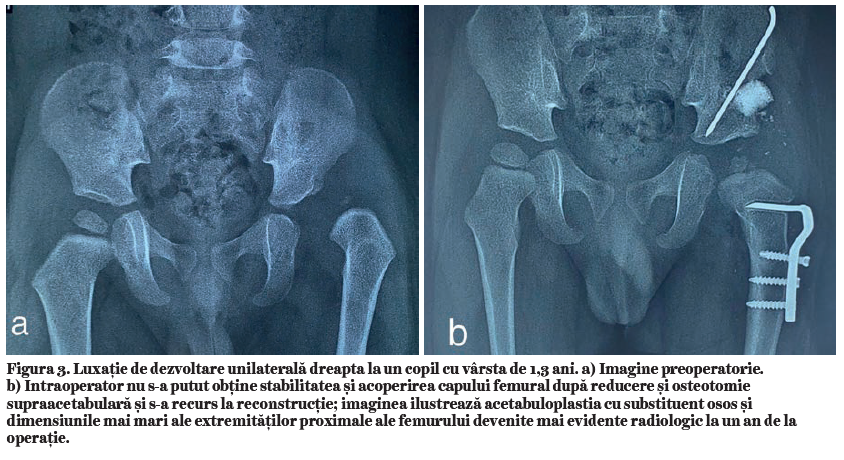
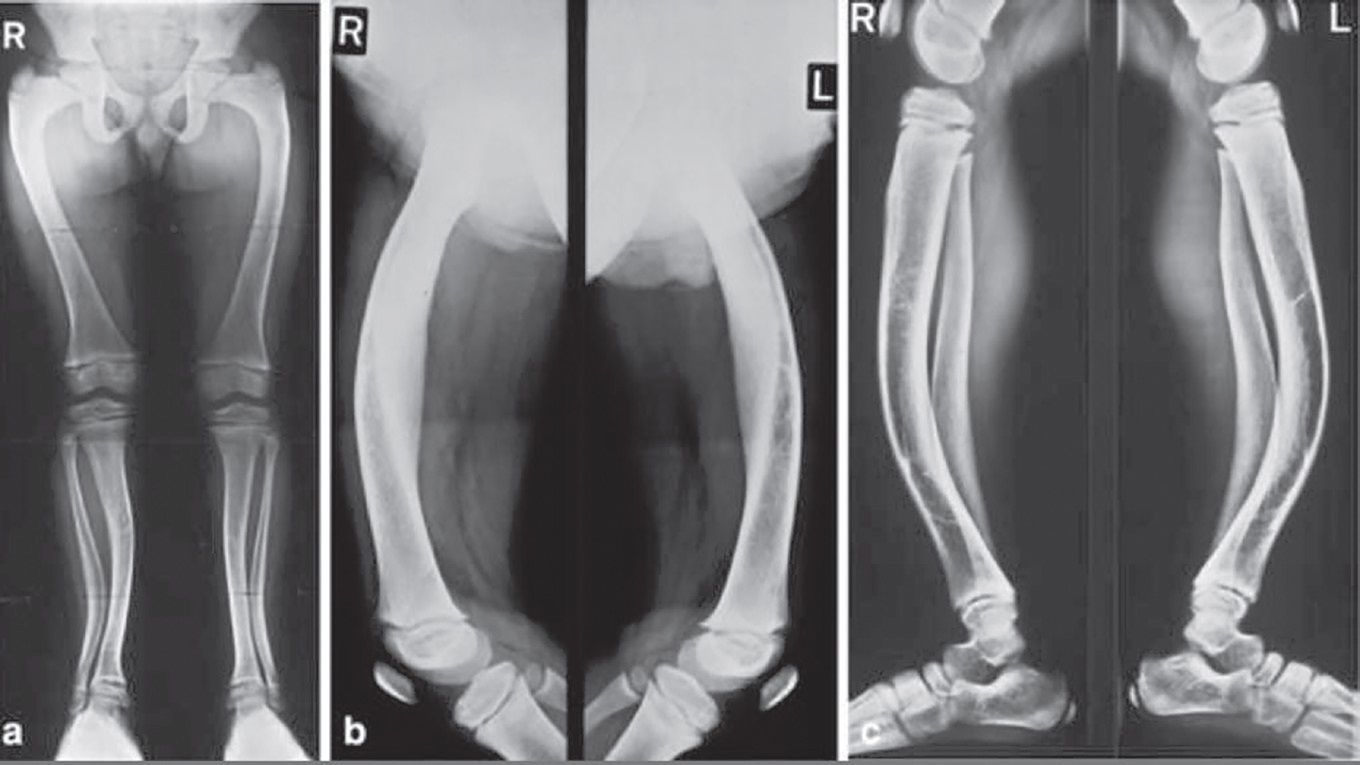
Simptomatologia este variată. Există forme ușoare, în care este prezentă numai hipofosfatemie, forme moderate, la care se adaugă și modificări osoase, și forme severe, în care apar modificări importante ale structurii osoase, ale configuraţiei oaselor lungi și deviaţii axiale de peste 30 de grade. Primele semne și simptome apar după ce copilul începe să meargă, când oasele membrelor sunt supuse greutăţii corpului în timpul ortostatismului și mersului. Astfel apar coxa vara, asociat cu femururi și tibii încurbate, genu var, genu valg sau deficitul staturo-ponderal, ca semn prevestitor al nanismului (fig. 2a și 2b, fig. 3).
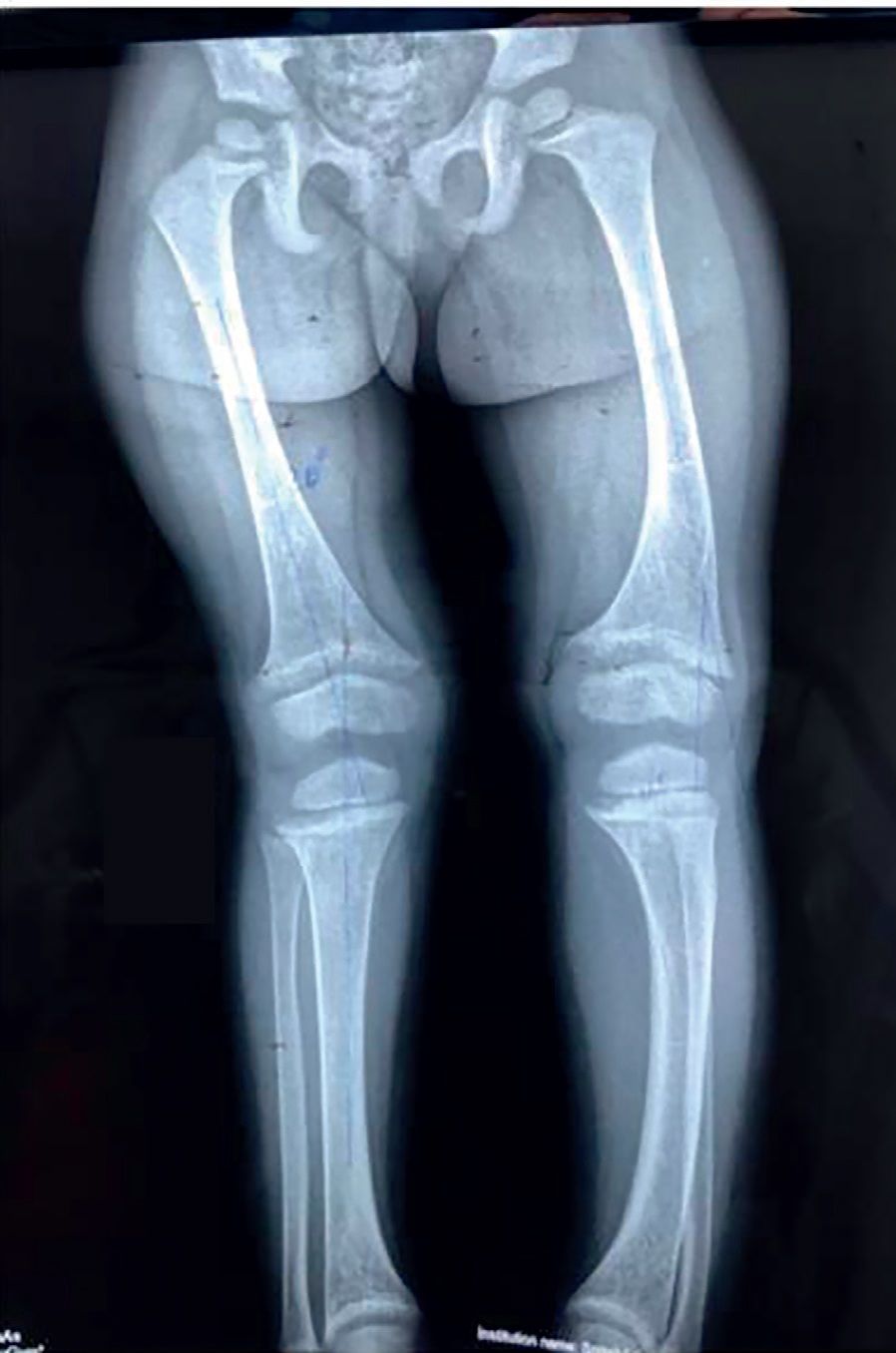

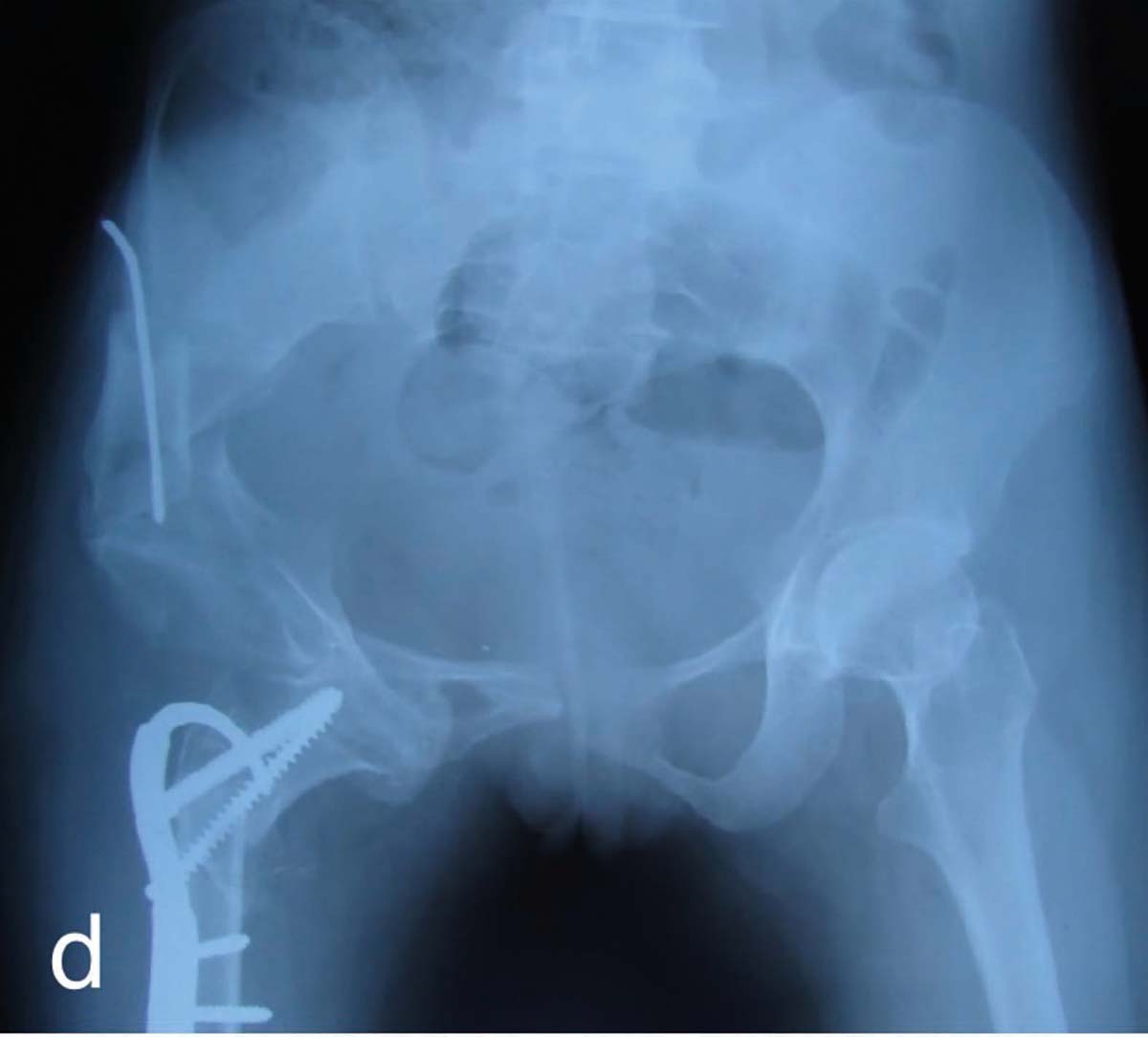
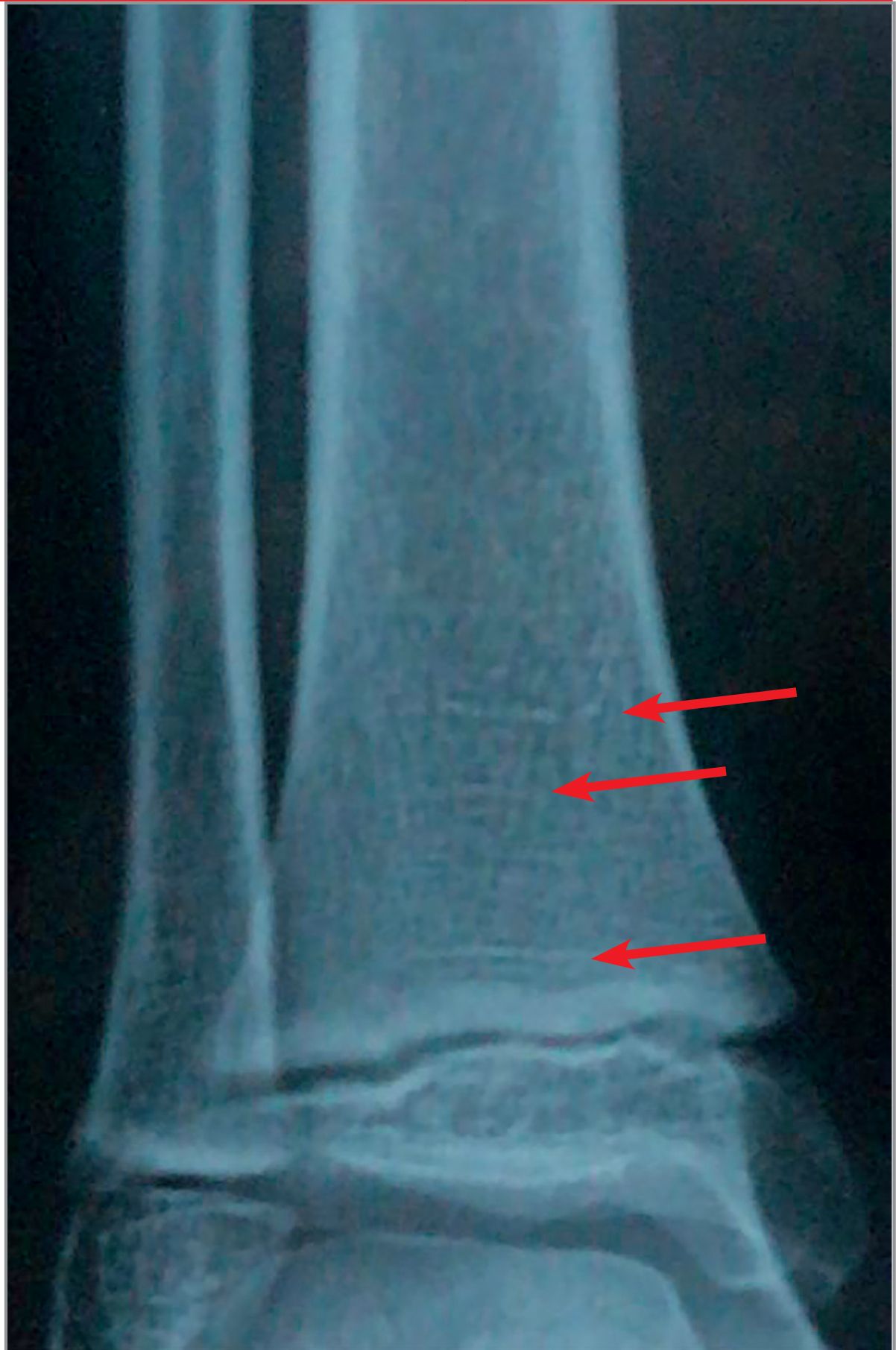
Alte simptome ce pot apărea în evoluţia bolii includ: durerile osoase, ca urmare a microfracturilor tramei osoase (fig. 4), hipotonii și dureri musculare, dureri articulare cauzate de osificări heterotopice, la nivelul tendoanelor și ligamentelor, mers legănat, desincronizat și dezvoltarea anormală a dinţilor. La adult pot apărea dureri articulare, abcese dentare, calcificări parţiale ale tendoanelor, ligamentelor și capsulei. Aceste calcificări parţiale dau frecvente entezite care afectează mersul.
Cele mai frecvente simptome, prezente în 80-99% dintre cazuri, sunt: smalţul dentar anormal, metafizele anormale, genu var, hipofosfatemia, luxaţiile articulare, osteomalacia, mătăniile costale și abcesele dentare. Alte simptome se găsesc în proporţii mai mici, la 30-79% dintre cazuri: craniosinostoza, entezitele, osteoartritele și nanismele. În 5-29% dintre cazuri apar fracturi recurente și afectarea auzului.
Examenul de laborator evidenţiază o fosfatemie scăzută, calcemia normală sau diminuată și o calciurie ușor sau moderat scăzută. Fosfataza alcalină este crescută. Mersul este șchiopătat și dizarmonic, cu desincronizări evidente între membrele pelvine și toracice. Pacienţii cu genu valg accentuat obosesc repede și adesea evită să meargă liberi pe distanţe lungi. Când deviaţiile axiale depășesc 30 de grade, mersul devine dizgraţios. Când deviaţia în valg este mai mare de 50 de grade, mersul nu mai este posibil. Modificările severe în var sau în valg dau copiilor o stare de disconfort în mers și uneori pot duce la fracturi spontane.
Creșterea este încetinită, iar încetarea ei se produce mai devreme, motiv pentru care la aproximativ 40% dintre pacienţi apare nanismul. Acești pacienţi nu depășesc talia de 140 cm. Evoluţia bolii se oprește odată cu încetarea creșterii, dar se poate redeschide după o sarcină sau după vârsta de 40-50 de ani.
Explorarea radiologică evidenţiază aspecte similare cu cele din rahitismul carenţial. Pe lângă încurbările oaselor lungi și deviaţiile axiale ale membrelor pelvine, apare o structură osoasă cu areole mai largi și travee osoase îngroșate. Nucleii osoși au o structură neregulată, sunt mai mici în raport cu vârsta, iar metafizele sunt lărgite și uneori neregulate. Ca urmare a evoluţiei bolii, în imediata apropiere a cartilajului de creștere, metafizar, apar benzi transverse de osificare. Pe conturul corticalei pot apărea exostoze. La anumiţi pacienţi sunt prezente imagini chistice sau semne ale unor fracturi. La nivelul rahisului, corpii vertebrali au structura ștearsă în special în segmentul lombar, devenind dreptunghiulari, iar platourile sunt ușor scobite. Pe radiografia craniului constatăm scafocefalia și închiderea precoce a suturilor.
Rahitismul pseudocarenţial este cea mai frecventă formă de rahitism genetic și prezintă leziuni scheletice asemănătoare cu ale rahitismului carenţial, diferenţiate în funcţie de severitatea bolii, ce pot debuta precoce sau tardiv. Este un rahitism vitamino-rezistent, cu dezechilibru homeostazic al fosfaţilor, determinat de pierderea fosfatului prin urină, ca în rahitismul hipofosfatemic, și care are specific hipofosfatemie și nivel normal sau scăzut de 1,25-dihidroxicolecalciferol.
Rahitismul Prader cu debut precoce este o formă de rahitism pseudocarenţial care debutează în primul an de viaţă și are o evoluţie gravă. Clinic, apar deformări severe ale membrelor, toracelui și rahisului. Frecvenţa fracturilor spontane depinde de încurbarea oaselor și severitatea bolii.
Analizele de laborator pun în evidenţă o hipocalcemie și o fosfatemie la limita inferioară a normalului. Uneori poate fi prezentă și aminoacidurie. Manifestările osteoarticulare sunt similare cu cele ale rahitismului carenţial. Clinic, apar segmente ale membrelor încurbate, în special coapsa și gamba, și deviaţii axiale.
Radiologic, trama osoasă este ștearsă, corticalele – subţiate, metafizele – lărgite, cartilajele de creștere – mai înalte și imprecis delimitate spre diafiză. Se pot pune în evidenţă linii de fractură consolidate în poziţie corectă sau consolidate vicios, în special la nivelul oaselor încurbate, acolo unde curbura prezintă maximumul de amplitudine. Nucleii epifizari apar cu întârziere, lungimea oaselor lungi este mai mică, iar talia unui număr mare de copii cu această afecţiune nu depășește 140 cm. Nanismul rahitic poate fi corectat chirurgical. Tratamentul cu doze mari de vitamina D, 6-8 g/zi, în unele cazuri, duce la o creștere normală sau cvasinormală.
Rahitismul Mc Cance cu debut tardiv este o formă de rahitism în care manifestările bolii apar în adolescenţă sau la vârsta adultă. Boala debutează la nivelul rahisului și membrelor. Deviaţiile axiale apar mai târziu, în special la nivelul membrelor și mai ales sub formă de genu var sau tibia vara. Adesea apar deformări ale rahisului sub formă de cifoză sau cifoscolioză.
Examenul radiologic evidenţiază o structură osoasă cu travee groase și rare, astfel încât alveolele devin foarte evidente. În formele severe, segmentele metafizare au transparenţa similară cu a părţilor moi, iar corticalele sunt mult subţiate sau dispărute. Pot apărea fracturi spontane, atât metafizar, cât și diafizar, care au caracterul fracturilor din sindromul carenţial Milkmann-Looser. Examenul biochimic arată o fosfatemie scăzută, calcemia normală și fosfatazele alcaline uneori crescute.
Tratamentul cu doze mari de vitamina D3 asociat cu administrarea de calciu și fosfor poate duce la vindecare. Acest tratament nu are efect asupra deviaţiilor axiale și încurbărilor. Ele se tratează chirurgical prin hemiepifizodeză, dacă deviaţiile apar în perioada de început a adolescenţei, sau prin osteotomii. Când apar inegalităţi de membre mai mici de 4-5 cm se fac epifiziodeze, iar în nanisme se recurge la alungirile de membre.
Dacă vrei să fii la curent cu tot ce se întâmplă în lumea medicală, abonează-te la „Viața Medicală”, publicația profesională, socială și culturală a profesioniștilor în Sănătate din România!
Titularii abonamentelor pe 12 luni sunt creditați astfel de:

Dacă vrei să fii la curent cu tot ce se întâmplă în lumea medicală, abonează-te la „Viața Medicală”, publicația profesională, socială și culturală a profesioniștilor în Sănătate din România!
Află mai multe informații despre oferta de abonare.
Cookie-urile ne ajută să vă îmbunătățim experiența pe site-ul nostru. Prin continuarea navigării pe site-ul www.viata-medicala.ro, veți accepta implicit folosirea de cookie-uri pe parcursul vizitei dumneavoastră.
Da, sunt de acord Aflați mai multe