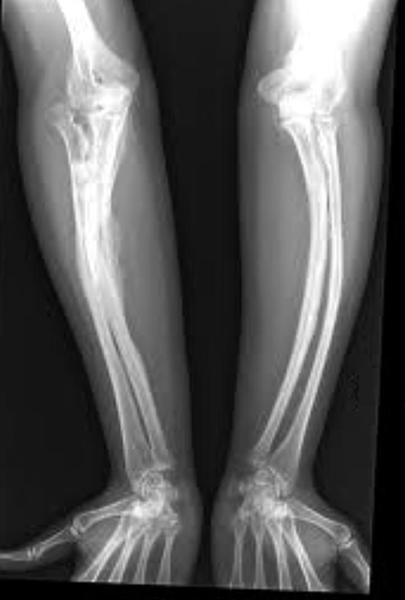
Osteogeneza imperfectă (OI) este o boală ereditară rară a ţesutului conjunctiv, caracterizată prin fragilitate osoasă. Iniţial s-a numit „boala oamenilor de sticlă“ sau „boala oaselor de sticlă“. Literal, OI specifică formarea imperfectă a oaselor sau osteoporoza congenitală. Ea este în general o anomalie de producţie a colagenului, principala proteină fibroasă din os. Putem compara rolul colagenului din os cu cu rolul fierului din betonul armat. Colagenul este de asemenea prezent în piele, tendoane, sclerotica ochiului şi în dentină (constituentul principal al dinţilor, împreună cu smalţul). Consecinţa cea mai cunoscută a OI este apariţia fracturilor multiple şi recurente fără traumatisme majore. Severitatea sa este foarte variabilă: fracturi prenatale şi deces perinatal sau forme foarte fruste. Tulburările extrascheletice sunt şi ele asociate în grade foarte variabile. În 1979, Sillence a propus o clasificare a OI în patru categorii, bazate pe criterii clinice; cu unele modificări minore, aceasta este încă utilizată. Nu este posibil întotdeauna să se determine din ce categorie face parte boala unui individ.
Osteogeneza imperfectă este o boală rară. Prevalenţa sa nu este cunoscută cu exactitate, dar este estimată la 1/10.000–1/20.000. Această estimare nu include formele moderate de boală, care pot trece inaparente. OI survine la toate populaţiile, fără predominanţă etnică sau rasială, şi la ambele sexe.
Doar 0,008% din populaţia globului este afectată de această boală (aproximativ 500.000 de persoane). (...)
Mai multe detalii în ziarul nostru, Viaţa medicală.
Clasificare
Semne clinice
Criterii de diagnostic
Diagnosticul diferenţial
Aspecte genetice
Sfatul genetic
Diagnosticul prenatal
Evoluţie şi prognostic
Posibilităţi de tratament, îngrijire şi urmărire
Viaţa cotidiană

.jpg)





