Sindromul Angelman (SA) este o boală genetică
rară, apărută prin lipsa contribuţiei materne a regiunii 15q11-q13 (de pe
cromozomul 15), care poate fi urmarea unei deleţii de novo în majoritatea cazurilor (60%), a unei disomii uniparentale
paterne (ambii cromozomi 15 de origine paternă) în mai puţine cazuri (7%), a
unor mutaţii în centrul de amprentare (2–5%) sau ale genei UBE3A (10%). Se
caracterizează prin retard mintal sever, retard sever de vorbire, facies
caracteristic, microcefalie, mişcări de tip ataxic, convulsii şi un
comportament particular, cu crize paroxistice de râs. Retardul de dezvoltare
devine evident în jurul vârstei de 6 luni, iar tabloul clinic caracteristic se
conturează adesea după vârsta de 1 an. Are o frecvenţă de 1:12.000-20.000 de nou-născuţi.
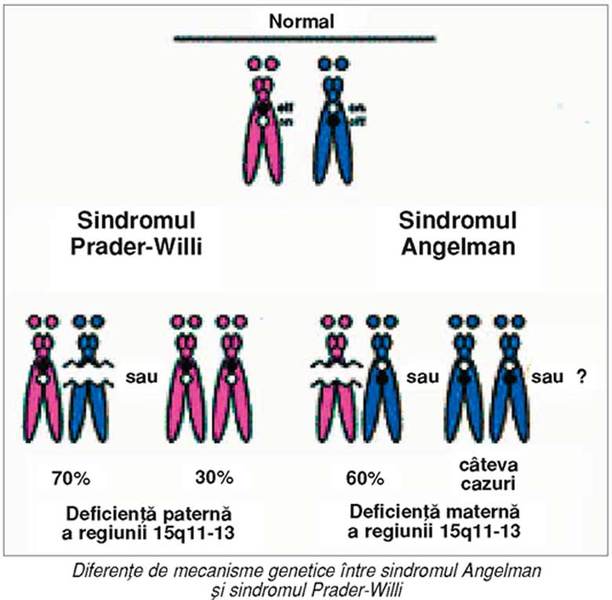
Aspecte genetice
Sindroamele Angelman şi Prader-Willi rezultă
prin deleţia unei regiuni a cromozomului 15, care conţine gene amprentate. Gena
implicată în sindromul Angelman este de origine maternă, astfel încât deleţia
acestei alele determină boala. Pe de altă parte, deleţia paternă a aceleiaşi
regiuni produce sindromul Prader-Willi, reflectând prezenţa uneia sau a multor
gene exprimate, de origine paternă, în această regiune.
Fenotipul particular al sindromului Angelman
este cauzat de pierderea contribuţiei materne din regiunea amprentată 15q11-q13
şi este atribuit lipsei de expresie sau funcţie a alelei materne UBE3A (ubiquitin protein ligase E3A).
Indivizii cu deleţii mari par a fi mai sever afectaţi în comparaţie cu cei cu
disomie uniparentală, cu un tablou clinic dominat de incidenţa crescută a
convulsiilor, microcefalie, hipopigmentare, retard sever al achiziţiilor
psihomotorii. Rearanjamente cromozomiale care implică regiunea critică
(translocaţie, inversie) se pot identifica la 1% din aceşti pacienţi.
Semne clinice
La sugar. Evoluţia prenatală a
pacienţilor cu SA, precum şi parametrii morfometrici la naştere sunt, de
obicei, în limite normale. Nou-născuţii cu sindrom Angelman pot avea dificultăţi
în alimentarea la sân sau cu biberonul (din cauza dificultăţii de supt specifică
sindromului şi a refluxului gastroesofagian) şi manifestă hipotonie musculară.
Sindromul este de obicei suspectat la vârsta la care copilul ar trebui să
înceapă să meargă, ca urmare întârzierii atingerii acestei etape şi a
întârzierii limbajului. Unii sugari au dispoziţie afectivă fericită cu chicotit
excesiv şi crize paroxistice de râs, comportament hiperkinetic cu spectrul atenţiei
scăzut. 50% dezvoltă microcefalie până la vârsta de 12 luni. Strabismul poate
fi evident. Pacienţii au limba protruzionată, cu tendinţă de salivare excesivă.
Mişcările ataxice se pot instala până la 12 luni, adesea cu reflexe
osteotendinoase exagerate.
La copilul mic (1–3
ani).
Crizele convulsive se instalează la vârsta de 1–3 ani şi pot fi tonico-clonice,
convulsii mioclonice, crize de absenţă, status
epilepticus şi status mioclonic major, precum şi crize minore.
Toate etapele de dezvoltare vor fi
întârziate, astfel mersul este realizat între 2,5 şi 6 ani şi este ţeapăn,
sacadat, cu membrele superioare în flexie şi pronaţie. 10% din copii nu merg
niciodată. Afectarea limbajului este severă. Este rară folosirea cuvintelor
potrivite într-o manieră care are sens. Limbajul receptiv este întotdeauna mai
dezvoltat decât cel expresiv.
Copiii de vârstă mai
mare şi adulţi. Sunt
capabili de comunicare prin arătarea obiectelor sau gesticulând şi folosind cartonaşe
de comunicare.
Instalarea pubertăţii are loc în mod
fiziologic şi atât bărbaţii, cât şi femeile sunt capabili de procreare.
Fertilitatea este aparent normală: în 1999 s-a raportat un caz de transmitere a
deleţiei la fetus de la mama afectată.
Adulţii tineri au sănătate fizică bună, cu
excepţia crizelor convulsive, a constipaţiei frecvente, iar scolioza se
accentuează cu vârsta, fiind raportată la 40% din adulţi, majoritatea femei. Cu
avansarea în vârstă, fenotipul se schimbă, fiind marcat de prognatism,
macrostomie şi buză inferioară proeminentă, iar protruzia limbii a fost
raportată atât la aduţii care prezintă crize convulsive, cât şi la cei fără
convulsii. Frecvenţa crizelor convulsive scade cu înaintarea în vârstă, dar 92%
din pacienţii adulţi fac crize convulsive. Cei care devin mai puţin activi au
tendinţa spre obezitate.
Date despre supravieţuire nu sunt raportate în
literatură, dar se pare că durata de viaţă este în limite normale; pacienţii nu
vor fi însă capabili niciodată să se întreţină singuri.
Diagnostic
Diagnosticul se bazează pe corelarea trăsăturilor fenotipice cu testele
moleculare şi/sau analiza citogenetică.
Clinic. Criteriile de
diagnostic ale sindromului Angelman au fost elaborate în acord cu comitetul ştiinţific
consultativ al Fundaţiei Americane a Sindromului Angelman. Profilurile
metabolic, hematologic şi biochimic sunt de obicei normale, iar structurile
corticale au aspect normal la evaluare IRM sau CT. Suspiciunea clinică necesită
o evaluare completă şi seriată a pacientului: consult neurologic – clinic,
paraclinic şi imagistic, oftalmologic, radiologic, psihologic, gastroenterologic.
Genetic. Tehnicile citogenetice
de înaltă rezoluţie trebuie să constituie primul pas în investigarea pacienţilor
diagnosticaţi clinic, pentru excluderea rearanjamentelor cromozomiale care
implică regiunea critică 15q11-q13 (vezi
tabelul). Analiza de metilare a ADN poate identifica mai mult de 80% din
pacienţi. Dacă rezultatul este normal, trebuie urmat de analiza secvenţială a
genei UBE3A. Dacă analiza de metilare este pozitivă, trebuie urmată de testul
FISH pentru confirmarea sau infirmarea microdeleţiei regiunii critice. Testul
FISH negativ necesită studiul polimorfismului ADN pentru diferenţierea disomiei
uniparentale de defectele de amprentare.
Sfatul genetic
Sfatul
genetic şi opţiunea testării genetice trebuie acordate tuturor familiilor în
care există un copil afectat de SA, indiferent de mecanismul de apariţie a
sindromului. Părinţii unui copil cu SA cauzat de deleţia regiunii critice sau
de disomia uniparentală 15 paternă au un risc mai mic de 1% de a mai avea încă
un copil afectat. Perioada optimă de determinare a riscului genetic şi de
prezentare a disponibilităţilor diagnosticului prenatal este înaintea sarcinii.
În cazul unui defect al centrului de amprentare sau a unei mutaţii în gena
UBE3A, riscul de recurenţă este de 50%. Sfatul genetic şi opţiunea testelor
genetice trebuie acordate şi rudelor mamei la care s-a identificat o mutaţie
UBE3A, o deleţie a centrului de amprentare sau un rearanjament cromozomial, dar
şi rudelor tatălui, în cazul disomiei uniparentale paterne. Dacă mama poartă o
mutaţie a centrului de amprentare, surorile ei ar putea să aibă aceeaşi mutaţie
şi acelaşi risc de 50% pentru un copil afectat cu sindrom Angelman. Fraţii ei
nu au risc pentru un copil afectat, dar pot transmite defectul fiicelor lor şi
pot avea nepoţi afectaţi.
Diagnosticul prenatal
Diagnosticul
prenatal este posibil pentru
identificarea tuturor mecanismelor patogenice, dar se indică după certificarea
defectului molecular al cazului index şi sfatul genetic corelat cu acesta. Se
pot preleva fie vilozităţi coriale (între săptămânile 10 şi 12 de sarcină), fie
lichid amniotic (între săptămânile 15 şi 18 de sarcină) şi se pot efectua atât analizele
citogenetice convenţionale şi FISH, cât şi metodele moleculare discutate
anterior, după extragerea de ADN fetal.
Pentru
familiile în care există un risc de recurenţă crescut (mutaţii ale UBE3A sau
deleţii ale IC), ar putea fi propus diagnosticul genetic  preimplantaţional, dar
există multe limitări din cauza hipometilării embrionare şi a numărului
restrâns de laboratoare care oferă aceste testări.
preimplantaţional, dar
există multe limitări din cauza hipometilării embrionare şi a numărului
restrâns de laboratoare care oferă aceste testări.
Diagnosticul diferenţial
Se face cu următoarele afecţiuni:
• suferinţă perinatală, encefalopatie
idiopatică, paralizie cerebrală, sindromul Lennox-Gastaut sau sindromul West
(caracteristici comune cu SA: retardul psihomotor şi paraplegia spastică);
• Sindromul Rett (prezintă adăugarea unei
evoluţii neurodegradante şi pierderea îndemânării);
• Sindromul Mowat-Wilson;
• Sindromul alfa-talasemie/retard mintal
X-linkat şi sindromul Phelan-McDermid (trăsături comune cu SA: retard mintal
sever, absenţa limbajului şi convulsii);
• Sindromul Prader-Willi (caracteristici
comune cu SA: hipotonia, masa musculară redusă).
Evoluţie şi
prognostic
Gradul de severitate a simptomelor asociate
sindromului Angelman este diferit de la un pacient la altul şi este corelat cu
mecanismul molecular cauzal. Durata de viaţă este una normală şi reproducerea
este posibilă. Terapia motorie şi logoterapia îmbunătăţesc considerabil
prognosticul. Evolutiv, o parte din trăsăturile sindromului
(hiperreactivitatea, convulsiile) pot diminua în intensitate şi frecvenţă.
Managementul pacienţilor
Hiperreactivitatea,
convulsiile, retardul mintal sever şi absenţa vorbirii sunt elementele clinice
majore care trebuie monitorizate.
• În perioada neonatală dificultăţile de
alimentaţie cauzate de hipotonie pot fi rezolvate prin utilizarea unor tetine
speciale. Refluxul gastroesofagian este o manifestare frecventă, dar poate fi
tratată prin poziţionarea sugarului sau cu medicaţie antireflux.
• Deoarece majoritatea cazurilor prezintă
crize convulsive la o vârstă precoce, acestea trebuie tratate corect, cu medicaţie
anticonvulsivantă.
• Unii pacienţi pot dezvolta probleme
vizuale, care ar trebui corectate deoarece interacţiunea vizuală este importantă
pentru minimizarea tendinţelor autistice şi automutilante pe care le pot
dezvolta pacienţii. Strabismul poate fi corectat chirurgical.
• Dieta bogată în fibre este recomandată
pentru managementul constipaţiei, iar atunci când aceasta este persistentă se
indică medicaţie laxativă.
• Salivarea excesivă este o trăsătură des
întâlnită la pacienţii cu retard mintal, adesea greu de tratat şi ar trebui
încercată terapia comportamentală. La ora actuală sunt posibile intervenţii
chirurgicale inovatoare, cu reimplantarea sau ligaturarea ductului salivar.
• Tratamentul ortopedic este necesar în
cazul scoliozei, care apare la o vârstă mai mare a copilului, pentru luxaţii
ale gleznei sau probleme ale tendonului lui Ahile cu anchiloze; în aceste
cazuri trebuie luate în considerare ortezele.
• Foarte importante sunt terapia fizică,
ocupaţională şi logopedia cu accent pe dezvoltarea modalităţilor non-verbale de
comunicare, incluzând metode ajutătoare precum cartonaşe. Deşi comunicarea este
foarte dificilă (maximum zece cuvinte), pacienţii agreează contactul cu alte
persoane, având o dorinţă deosebită pentru joacă şi interrelaţionare.
• De obicei, copiii au un retard neuromotor
sever, al cărui grad este important de evaluat pentru a se institui ulterior un
program de recuperare corespunzător. Achiziţiile sunt lente, dar printr-un
program susţinut acestea se menţin.
• Tratamentul insomniilor poate fi eficient
realizat prin administrarea de melatonină, care are rezultate după câteva săptămâni
de la administrare.
• Şcolarizarea trebuie să fie
particularizată şi flexibilă.