Boala
coronariană (BC) şi manifestările ei, inclusiv infarctul miocardic (IM)
sunt principala cauză de morbiditate şi deces în întreaga lume. Aceste afecţiuni
rezultă prin stenoza sau ocluzia arterelor coronare, care reduc sau blochează
fluxul sanguin, prin arterele afectate, spre miocard, producând ischemie sau necroză.
Cauza principală care determină îngustarea
unei artere coronare este placa
aterosclerotică iar ocluzia coronariană se produce prin formarea unui trombus pe ruptura plăcii. Ipotezele
care explică iniţierea, progresia şi ruptura plăcii aterosclerotice coronariene
au următoarele puncte-cheie şi etape (6): • disfuncţia endotelială • acumularea
de lipoproteine (în special LDL-colesterol) în „intima“ subendotelială • răspunsul
imun şi inflamator al arterei (infiltrarea peretelui vascular cu monocite şi limfocite)
• transformarea monocitelor în macrofage, care fixează lipoproteinele şi
formează „celule spumoase“, o caracteristică majoră a aterosclerozei precoce • formarea
unui miez necrotic de resturi
celulare şi colesterol • migrarea şi proliferarea celulelor musculare netede
vasculare, care produc un înveliş fibros în jurul miezului necrotic. Se formează
astfel „placa aterosclerotică“, care creşte treptat spre lumen şi produce
stenoza coronarei; uneori, peretele luminal al plăcii se subţiază şi se rupe, determinând
activarea plachetară şi formarea unui trombus, ce blochează fluxul sanguin şi
produce infarctul miocardic (IM). A devenit o certitudine că ateroscleroza (ATS) este o afecţiune inflamatorie progresivă care stă la baza BC (9).
Descifrarea factorilor, căilor şi mecanismelor moleculare ale aterosclerozei
reprezintă deci soluţia prevenţiei BC şi baza intervenţiilor terapeutice.
Studiile epidemiologice prospective
„clasice“ (Framingham, 1960; Helsinki, 1970, ş.a.) au evidenţiat o serie de factori de risc „tradiţionali“ pentru dezvoltarea BC (6, 7, 10). Unii dintre aceştia
– concentraţia lipidelor plasmatice (în special hipercolesterolemia),
hipertensiunea arterială (mai ales creşterea TA sistolice), fumatul,
hiperglicemia (în principal urmare a diabetului zaharat), obezitatea,
sedentarismul, stresul ş.a. – sunt factori de risc dobândiţi în cursul vieţii, pot fi evitaţi şi sunt modificabili.
Alţi factori de risc – vârsta (60–65 de ani) şi sexul (bărbaţii dezvoltă BC la
circa 60 de ani, cu zece ani mai devreme decât femeile) sau istoricul familial
sunt factori ficşi, ireversibili. Deşi s-a stabilit că istoricul familial este un
factor de risc independent pentru BC şi în special pentru IM prematur (<
50 de ani la bărbaţi şi < 60 de ani la femei), el nu a fost inclus în
algoritmii de predicţie a riscului cardiovascular (de exemplu, în scorul de
risc Framingham). Totuşi, concluzia acestor studii a fost că etiologia BC este complexă, multifactorială,
implicând combinaţii particulare între factori genetici multipli (poligenie) –
care determină o predispoziţie genetică
la boală – şi numeroşi factori de mediu.
Studiile familiilor şi gemenilor cu BC şi IM
au stabilit că aceste afecţiuni au o componentă genetică semnificativă: cota de
participare a eredităţii (heritabilitatea)
în etiologia BC a fost evaluată între 40% şi 60%, dar tinde să scadă cu vârsta
(6, 10); astfel, în IM prematur, heritabilitatea este 63%. Studiul descendenţilor
Framingham („Framingham offspring study“)arată că persoanele care au un părinte cu IM prematur prezintă un risc
dublu de boală coronariană comparativ cu indivizii fără istoric familial. O
parte din heritabilitatatea riscului de IM este independentă de alţi factori de risc, care, la rândul lor, au o
componentă ereditară importantă. În prezent, părerea generală este că jumătate din riscul de BC este reprezentat
de predispoziţia genetică (5), dar mecanismele sale sunt neclare şi
identificarea genelor de risc – dificilă.
Primele încercări de a rezolva „misterul“
geneticii BC s-au bazat pe studiul bolilor monogenice (mendeliene) ale metabolismului lipidelor. Prototipul
acestor studii este hipercolesterolemia
familială (HF) – o boală genetică caracterizată prin creşterea
LDL-colesterolului (LDL-C), care se depune în vase şi alte ţesuturi; HF se
transmite autozomal-dominant, heterozigoţii (An) fiind mai numeroşi (prevalenţa
de 1:500) dar moderat afectaţi comparativ cu homozigoţii (AA), foarte rari (1
la un milion) dar cu forme grave şi precoce de ATS. Analiza fibroblaştilor de
la bolnavii cu HF a evidenţiat un defect de fixare a LDL-C şi a dus la
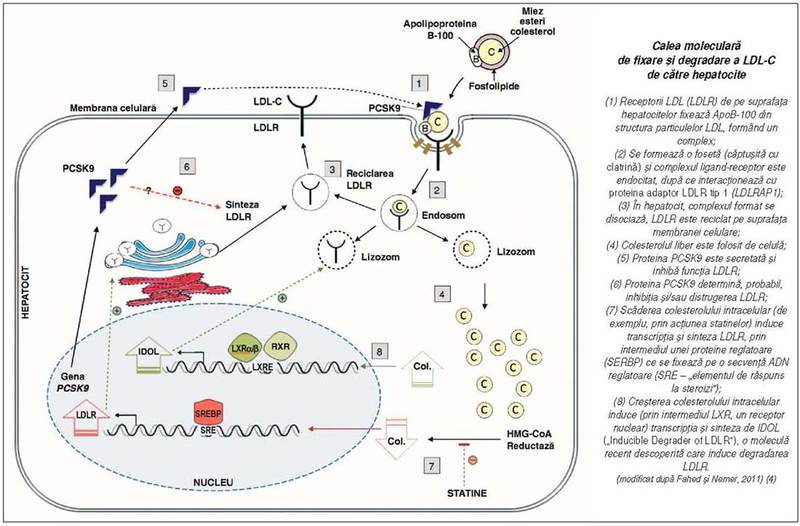
identificarea receptorilor pentru LDL (v.
figura). Mutaţiile cu pierderea funcţiei genei LDLR (ce codifică receptorii de LDL) explică transmiterea dominantă
a bolii în familii şi efectul dependent de dozajul genic (homozigoţii AA fiind
mai grav afectaţi decât heterozigoţii An).
Ulterior, studiul genetic al unor familii cu
hipercolesterolemie familială a dus la descoperirea altor mutaţii dominante
cauzale în genele: APOB (codifică
apolipoproteina B-100, componentă a particulelor de LDL-C)1 şi PCSK9 (codifică proprotein-convertaza
subtilin/kexină tipul 9) (v. figura);
aceste mutaţii reduc fixarea LDL-C la receptori (4). A fost identificată şi o
formă de hipercolesterolemie recesivă produsă de mutaţii în gena LDLRAP1 (codifică proteina 1 adaptor al
receptorului LDL)2. Toate aceste mutaţii
au confirmat şi dezvoltat calea moleculară
pentru fixarea şi degradarea LDL-C în celule, descoperită de Brown şi
Goldstein (Premiul Nobel, 1985), care elucidează mecanismele hipercolesterolemiei.
Autorii au meritul de a fi introdus trei concepte fundamentale pentru biologia
celulară: endocitoza mediată de receptori, reciclarea receptorilor şi reglarea
feedback a sintezei receptorilor. Ultimul concept stă la baza mecanismului prin
care statinele scad selectiv nivelul LDL-C plasmatic, reducând riscul de IM,
prin creşterea sintezei receptorilor LDL (statinele inhibă HMG-CoA reductaza ce
acţionează în primele etape ale sintezei colesterolului; scăderea nivelului
colesterolului în hepatocite va creşte transcripţia genei LDLR; v. figura). În
felul acesta, geneticienii au adus o nouă contribuţie valoroasă la înţelegerea
patogeniei BC. De menţionat că studiile lipoproteinelor plasmatice au
demonstrat că prin creşterea HDL-colesterolului (HDL-C) scade riscul
cardiovascular. Trialuri clinice cu agenţi care cresc HDL-C şi au efecte
antiaterosclerotice şi antiinflamatoare sunt în curs de desfăşurare (6).
O altă abordare „pregenomică“ pentru
identificarea genelor implicate în producerea susceptibilităţii la formele
multifactoriale de BC se bazează pe studiile de înlănţuire şi de asociere genică,
care au ca scop stabilirea unei asocieri semnificative a unei anumite regiuni
cromozomiale (numită generic locus)
sau a unei variante alelice a unei gene cu BC. Analizele de înlănţuire genică s-au efectuat pe familii de bolnavi
cu BC/IM prematur, pe baza prezumţiei că bolnavii vor avea mai frecvent în
comun o anumită variantă genetică comparativ cu persoanele sănătoase din aceeaşi
familie (3). Studiile câtorva sute de familii cu IM prematur au stabilit că
gena LRP6 (LDL receptor-related protein 6) ar putea fi un candidat credibil
pentru o formă rară de IM transmis autozomal dominant3, iar variante ale
genelor ALOAX5AP (ce codifică
proteina activatoare a 5-lipooxigenazei, implicată în producerea de leukotriene) şi LTA4H (ce codifică hidroxilaza leukotrienei 4) se asociază cu un
risc crescut de IM; în plus, aceste observaţii sugerează că activitatea
leukotrienelor, implicate în procesele inflamatorii, poate contribui la
fiziopatologiei BC (7, 10).
Studiile
de asociere genetică se efectuează într-o populaţie şi compară incidenţa
unor anumite variante alelice ale unor gene
candidat (implicate în căi metabolice sau factori de risc cunoscuţi pentru
BC) la bolnavi, cu incidenţa lor la un grup control. Majoritatea datelor
furnizate de studiile „caz-control“ (de exemplu, implicarea genelor pentru
conexină, trombospondină etc.) nu au fost confirmate prin studii replicative
(explicaţii tehnice, statistice, etnice ş.a.) (7, 10). O singură excepţie o
constituie gena ABO (ce codifică o glicozil-transferază implicată în
sinteza antigenelor de grup sanguin), care contribuie semnificativ la riscul de
IM; alelele dominante „A“ şi „B“, comparativ cu „0“, au fost asociate cu
tromboembolismul şi cu IM (dar nu şi cu angina pectorală) încă de acum 50 de
ani. Alela recesivă „0“ este lipsită de activitate enzimatică şi studii recente
au evidenţiat asocierea sa cu reducerea concentraţiei plasmatice a unor factori
ai coagulării (factorul VIII şi factorul von Willebrand), sugerând că asocierea
genei ABO cu IM reflectă cel puţin un
rol parţial în modularea trombozei în ATS (7). În contrast cu analizele de
asociere pentru BC, studiile genelor candidat implicate în metabolismul
lipoproteinelor au avut mai mult succes, demonstrând asocieri semnificative cu
BC şi IM a unor variante ale genei LPA,
ce codifică lipoproteina(a) şi a
genelor ce codifică apolipoproteinele E şi
A5.
În concluzie, înainte de finalizarea
Proiectului Genomul Uman (2001), studiile de identificare a genelor
responsabile pentru heritabilitatea BC şi
IM au permis descoperirea unor variante genetice rare, la puţini indivizi,
care au început să elucideze „misterul genetic“ al patogeniei BC, permiţând noi
abordări terapeutice eficiente (de exemplu, introducerea terapiei cu statine).
Cu toate acestea, complexitatea genetică a aterosclerozei nu a fost intuită.
„Explozia“ cunoştinţelor în domeniul
geneticii/genomicii BC s-a produs însă în ultimii cinci ani, datorită
progreselor înregistrate în secvenţierea genomului uman şi identificarea unui
număr foarte mare de polimorfisme ADN, în special SNP („polimorfismul unui singur nucleotid“). Aşa cum am discutat în
articolul anterior (3), în 2005 au început studiile
de asociere utilizând întregul genom (în engleză „genome wide association studies“ – GWAS); aceste studii de tip „caz-control“ permit determinarea unor
„variante comune (prezente la > 5% din populaţie) – asociate cu boli
comune“, folosind genotiparea concomitentă (pe microreţele ADN) a sute de mii
de markeri SNPs (distribuiţi în tot genomul), la zeci de mii de indivizi
dintr-o populaţie. Markerii SNPs index asociaţi semnificativ statistic cu boala
definesc o regiune cromozomială în care se află varianta unei gene de
susceptibilitate la boală. Identificarea genei cauzale şi determinarea funcţiei
şi mecanismului prin care această genă participă la realizarea bolii necesită,
evident, alte studii.
Aplicarea studiilor de asociere la nivelul întregului
genom în bolile cardiovasculare şi în special la BC a fost un succes, determinând
progrese remarcabile în analiza factorilor de risc „tradiţionali“, dar şi a
unor factori noi, în înţelegerea mecanismelor patogenice şi în aplicarea cunoştinţelor
la îmbunătăţirea tratamentului (inclusiv pe baza genotipului individual). Menţionăm
că studiile de farmacogenomică au
identificat variante genetice ce influenţează răspunsul individual la unele
medicamente folosite frecvent în terapia bolilor cardiovasculare: warfarină,
clopidogrel, statine, beta-blocante, vasodilatatoare ş.a., despre care am
discutat într-un articol anterior (2).
În perioada 2007–2011 s-au efectuat 15
studii GWA şi au fost publicate (în 2011) două meta-analize4 care au vizat: • BC
(confirmată angiografic) şi IM • IM prematur (< 50 de ani la bărbaţi şi <
60 de ani la femei) • factorii de risc majori modificabili (dislipidemii, HTA,
DZ tip 2) şi obezitatea • BC „subclinică/presimptomatică“ (ce precede cu câţiva
ani debutul simptomelor), definită prin markeri biochimici (fibrinogen,
proteina C reactivă, molecula de adeziune intercelulară 1, homocisteina
plasmatică) sau măsurători imagistice (îngroşarea intimei şi mediei carotidei,
plăcile carotidiene).
Studiile GWA au identificat circa 30 de regiuni cromozomiale (loci) asociate semnificativ cu BC şi IM.
Unele dintre acestea conţin gene cunoscute a fi implicate în metabolismul lipidelor şi procesele
inflamatorii care conferă risc la BC – cum ar fi genele LDLR, PCSK9, LPA, APOA1, APOB – confirmând
importanţa LDL-colesterolului în ATS. Alte regiuni genomice conţin secvenţe
reglatoare şi/sau „gene-candidat noi“ (ADAMTS7,
LIPA, CELSR1 ş.a.), al căror rol în ATS nu este cunoscut încă (6, 7, 10).
GWAS identifică gene „scoase din context“, fără informaţii despre funcţia lor,
informaţii ce pot fi dobândite numai prin alte studii pe modele celulare sau
animale. De exemplu, un locus (1p13) – din regiunea 13 a braţului scurt al
cromozomului 1 – cu efect puternic asupra IM şi LDL-C, conţine trei gene (CELSR2, PSRC1 şi SORT1) despre care nu se ştia anterior că participă la metabolismul
lipidelor (7). Studii funcţionale ample pe culturi de ţesuturi umane şi pe şoarece
au stabilit că gena SORT1 codifică o proteină – sortilina
– implicată în secreţia hepatică de VLDL, care, prin lipoliză extrahepatică,
formează LDL. O variantă comună a unui situs de reglare a genei a SORT1 creşte transcripţia şi producţia
de sortilină, scăzând astfel secreţia de VLDL, nivelul plasmatic al LDL-C şi
riscul de IM. Această nouă cale de reglare a metabolismului lipoproteinelor va
avea foarte probabil consecinţe terapeutice benefice.
Cel mai puternic locus asociat cu BC şi IM
prematur se află în regiunea 9p21,
care, surprinzător, nu conţine gene care să fi fost asociate anterior cu BC sau
cu factorii de risc la BC (7, 10). Cercetătorii au încercat să elucideze
mecanismul prin care această regiune genomică poate contribui la ATS. Astfel, a
fost identificată o secvenţă ce codifică o moleculă de ARN lung, numită ANRIL
(de la „antisense noncoding RNA in the INK4 locus“)5 ce controlează expresia
genelor CDKN2A/2B şi ARF în endoteliul vascular6, gene
implicate în reglarea ciclului celular. Un alt studiu a identificat – în aceeaşi
regiune – un element reglator (activator sau „enhancer“) al expresiei genei IFNA21,
care codifică interferonul gama în celulele endoteliale, implicată în căile
inflamatorii ale ATS. Rolul regiunii 9p21 în BC şi IM rămâne încă neelucidat.
Despre genomica ATS şi implicit a BC ar mai
fi de subliniat două aspecte:
• Studiile GWA asupra aterosclerozei
precoce, evaluată ultrasonografic prin măsurarea grosimii intimă-medie a
carotidei şi plăcilor carotidiene au evidenţiat câţiva loci asociaţi cu aceste
fenomene; dar numai o singură genă (EDNRA,
ce codifică receptorul endotelină tip A) este asociată şi cu BC, precum şi cu
HTA. Ceilalţi loci, neasociaţi şi cu BC, sunt probabil specifici tipului de
arteră (arcul aortic, carotidele) sau stadiului precoce al bolii (6).
• BC afectează bărbaţii la o vârstă mai tânără
decât femeile, fapt ce sugerează rolul posibil al unor variante genetice
localizate pe cromozomul Y. Acest fapt a fost confirmat recent de cercetătorii
britanici, care au identificat pe cromozomul Y un haplogrup I, ce produce – la
persoanele care îl posedă – un risc de BC cu 50% mai mare comparativ cu bărbaţii
care au alte haplotipuri; acest risc este independent de factorii de risc tradiţionali
şi socioeconomici. Analiza expresiei genelor arată că indivizii cu haplogrupul
I au o dereglare a sistemului imun, afectând în egală măsură autoimunitatea şi
inflamaţia. Se poate presupune că predispoziţia crescută a bărbaţilor la BC
depinde de… moştenirea pe linie paternă (1).
Alte studii de asociere la nivelul întregului
genom au vizat factorii de risc ai BC şi
în special lipidele plasmatice7. Cel mai amplu studiu realizat de Global Lipids Genetic Consortium – GLGC
(2010) a fost un screening genomic, la peste 100.000 de persoane de origine
europeană, pentru a identifica variante comune asociate cu cel puţin una din
lipidele plasmatice: colesterol total (CT), LDL-colesterol (LDL-C),
HDL-colesterol (HDL-C) şi trigliceride (TG). Prin acest studiu (12), s-au
identificat 95 de loci (din care 58 noi!) asociaţi semnificativ cu
lipoproteinele plasmatice. Mulţi dintre aceştia se află în/lângă gene ce
codifică reglatori lipidici cunoscuţi (de exemplu, LDLR, PCSK9, APOB, APOE, CETP, HMGR ş.a.), dar şi loci noi, validaţi
prin studii funcţionale. Este important de subliniat că unii dintre aceşti loci
conţin variante comune cu efecte mici în BC, dar şi mutaţii rare, cu efecte
mari, care produc bolile lipidice mendeliene.
Studiile GWA au permis înţelegerea mai
profundă a căilor metabolismului lipoproteinelor şi identificarea unor noi
oportunităţi terapeutice pentru BC. Un exemplu edificator îl constituie
identificarea (la circa 3% din persoane sănătoase de origine africană) unei
variante în gena PCSK9 (o alelă nulă,
mutaţie cu pierdere de funcţie8),
care, în stare heterozigotă (Na),
este asociată cu scăderea nivelului LDL-C (cu circa 30%) şi a riscului de BC şi
IM (cu circa 90%!) (11); s-au găsit şi persoane homozigote (aa, cu ambele gene mutante) care sunt sănătoase (!) şi care (prin
creşterea numărului LDL-R) au un nivel de colesterol (14–16 g/dl) mai mic decât
cel obţinut cu statine. Pe această bază s-a creat un anticorp monoclonal ce
blochează capacitatea proteinei PCSK9 normale de a distruge LDL-R, scade
nivelul LDL-C (rezultatele publicate în 2012, pe un trial de fază clinică 1,
sunt excelente) (11). Probabil că, după statine, PCSK9 reprezintă „un nou
capitol captivant“ în istoria colesterolului, dar nu şi ultimul: identificarea
recentă a proteinei IDOL – care, la fel ca PCSK9, induce degradarea LDL-R (v. figura) va genera un nou tip de
inhibitori ai colesterolului.
Cu toate rezultatele deosebite în
identificarea genelor implicate în căile patogenice ale ATS şi a ţintelor potenţiale
pentru noi intervenţii terapeutice, studiile GWA actuale au limite şi nu sunt
lipsite de critici, la care ne-am referit în articolul anterior (3). Genele de
susceptibilitate la BC şi IM identificate până în prezent explică numai o parte
din agregarea familială şi heritabilitatea
bolii iar valoarea lor predictivă pentru riscul cardiovascular (chiar prin
aplicarea unor „scoruri genotipice“), în ciuda entuziasmului iniţial, este
modestă. Aşadar, avea dreptate un cardiolog care acum zece ani scria: „Vă sfătuiesc
să mâncaţi mai puţine grăsimi sau, mai bine, să mâncaţi mai puţin; întâmplător,
fumatul este rău şi nici sedentarismul nu este bun. Trebuie să beţi mai puţin
de trei sticle de bere pe zi sau cel puţin să schimbaţi berea cu vinul roşu. Nu
uitaţi să vă luaţi medicaţia antihipertensivă. Acestea sunt sfaturile pe care
le dau obişnuit pacienţilor mei şi mă întreb de ce avem nevoie de teste
genetice?“ (5).
Pentru a depăşi aceste limite, „al doilea
val“ al studiilor GWA abordează variantele rare (sub 1%) dar cu efecte mari,
alte variante structurale în afară de SNPs (de exemplu, CNV – copy number variations – şi indels – insertions/deletions), utilizarea
secvenţierii integrale a genomului, analiza unor fenotipuri intermediare şi a
efectelor funcţionale ale genelor, precum şi studiul interacţiunilor dintre
gene („epistazie“) şi între factorii
genetici şi cei de mediu. Acest ultim aspect poate fi demonstrat de cercetările
recente privind efectele fosfolipidelor oxidate asupra endoteliului vascular
(6). Studiile GWA la om şi cercetările experimentale la şoarece au evidenţiat
asocierea puternică a oxidului de trimetilamină (TMAO) cu ateroscleroza. TMAO
rezultă prin oxidarea enzimatică a trimetilaminei (TMA), un gaz produs în
intestin prin acţiunea bacteriilor intestinale asupra colinei; TMAO contribuie
la formarea „celulelor spumoase“ (macrofage pline cu colesterol) în peretele
vascular. Nivelul TMAO şi acţiunea sa aterosclerotică depind de factori
multipli: dietă, variaţii în compoziţia florei intestinale şi variaţii în
expresia enzimei flavin-monooxigenază, care transformă TMA în TMAO (6).
Studiile GWA au arătat că mai avem multe de învăţat
despre căile şi mecanismele care stau la baza ATS şi BC; multe dintre regiunile
cromozomiale (locii) şi genele asociate puternic cu aceste afecţiuni nu par a
fi conectate cu căi patogenice cunoscute. Cert este că abordarea genomică a BC
este o mare speranţă pentru descifrarea mecanismelor sale patogenice şi, pe
această bază, pentru identificare unor noi ţinte terapeutice.
1Defectele familiale în
apolipoproteina B-100 au o prevalenţă de 1:1.000 indivizi.
2Gena LDLRAP1 mai este numită şi ARH de la „autosomal recesive hypercolesterolemia“.
3O altă genă, MEF2A, implicată iniţial în IM autozomal
dominant, nu a fost validată de studii ulterioare.
4CARDIoGRAM – „the Coronary ARtery DIsease Genome Wide Replication And Meta-analysis“ – studiu de tip
„caz-control“ pe > 150.000 de subiecţi de origine europeană – şi Coronary Artery Disease (CAD) Genetic Consortium.
5Simbolul oficial este
în prezent CDKN2B-AS1.
6Regiunea 9p21 a fost asociată şi cu alte fenotipuri vasculare
patologice (anevrisme de aortă sau artere intracraniene, boală arterială
periferică), sugerând că variaţii ale ADN din această regiune interferează
dezvoltarea ţesutului vascular (3).
7Studiile GWA în HTA şi
DZ tip 2, alţi doi factori majori de risc, au dus, de asemenea, la descoperirea
unor loci de susceptibilitate la BC.
8Gena PSCK9 codifică o proteină care împiedică
reciclarea LDL-R şi produce distrugerea lor în hepatocite (v. figura). Mutaţiile genei PCSK9
cu câştig de funcţie produc o formă de hipercolesterolemie familială.




.jpg)


