Descoperirea
multitudinii şi diversităţii variaţiilor
structurale ale genomului uman (SNPs, indels,
CNVs ş.a. – descrise în articolul precedent), care, prin configuraţia lor
specifică, determină individualitatea
genetică a fiecărei persoane, a deschis calea medicinii personalizate. Numeroase studii, realizate după
finalizarea (în 2004) a Proiectului Genomul Uman (PGU), au demonstrat că aceste
variaţii au o contribuţie semnificativă la evidenţierea diferenţelor
individuale de răspuns la agresiunile mediului, susceptibilităţii la bolile
comune ale adultului şi eficienţei acţiunii medicamentelor. Translaţia şi
implementarea cercetărilor experimentale în clinică, la patul bolnavului,
necesită însă depăşirea a patru obstacole majore:
• Identificarea şi catalogarea completă a tuturor tipurilor de variaţii structurale
(frecvente şi rare) în populaţii diferite;
• Corelarea variantelor specifice sau a
unor combinaţii ale acestora cu riscul diferit la bolile comune şi eficienţa
unor intervenţii terapeutice;
• Secvenţierea
genomului personal, mai rapidă şi la un cost acceptabil, pentru a stabili profilul individual al variantelor
structurale genomice;
• Interpretarea „în timp real“, de către
medici, a informaţiilor privitoare la structura genomului pacienţilor, pentru a
stabili fie măsuri adecvate de prevenire a bolii la persoanele cu risc, fie o
schemă terapeutică individualizată (cu maximum de beneficii şi minimum de
efecte adverse).
Primele
trei acţiuni au fost deja abordate, prin colaborarea amplă a cercetătorilor din
diferite ţări, şi se speră că în 10–15 ani rezultatele vor fi operaţionale în
medicina clinică; ele trebuie dublate de o pregătire susţinută a tuturor
practicienilor, pentru a deveni capabili să aplice progresele medicinii
genomice în folosul pacienţilor.
Identificarea şi cartografierea variaţiilor
structurale din genomul uman a început în 2002 odată cu lansarea proiectului internaţional HapMap. Obiectivul principal al
proiectului a fost realizarea unei hărţi
haplotipice a genomului uman, care să descrie modelele comune ale distribuţiei
variantelor structurale ce interesează un singur nucleotid („single nucleotid polymorphism“ – SNP), cu scopul facilitării
cercetărilor care urmăresc identificarea variantelor structurale ce afectează sănătatea,
boala şi răspunsul organismelor la medicamente şi factori de mediu. Soluţia cea
mai bună pentru stabilirea acestor corelaţii ar fi fost secvenţierea completă a
genomului la persoane cu şi fără o anumită boală dar, în 2002, această soluţie
nu era posibilă din cauza costurilor ridicate şi timpului îndelungat pentru
secvenţierea întregului genom. Proiectul HapMap
propunea o „scurtătură“, studiind numai SNPs frecvente, ale căror alele se întâlnesc
la cel puţin 5% din populaţie.
Pentru
a înţelege logica proiectului HapMap,
să ne reamintim că, în populaţie, la persoane diferite, un acelaşi segment de
ADN dintr-o anumită regiune cromozomială poate prezenta, în anumite situsuri,
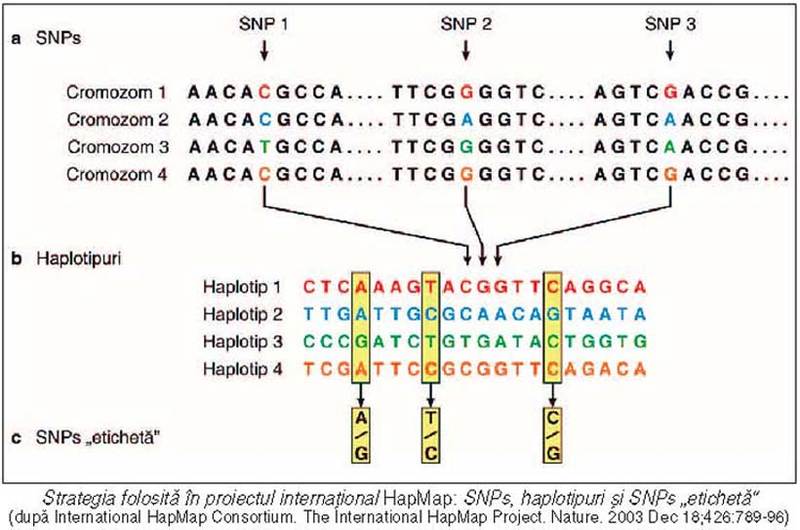
două variante („alele“) ale unui singur nucleotid (SNPs). De exemplu, în secvenţa
nucleotidică din figura alăturată (a)
trei nucleotide diferă de la o persoană la alta, prezentând alelele C/T, G/A şi G/A, celelalte
nucleotide fiind identice. S-a stabilit că alelele unor SNPs vecine, situate
liniar pe un singur cromozom din perechea de omologi, sunt corelate, formând o combinaţie
specifică, care se transmite împreună, „în bloc“, de la părinţi la descendenţi,
numită haplotip. În aceeaşi figură (b) sunt prezentate 20 de SNPs comune
(care se găsesc intercalate într-o secvenţă de ADN de circa 6.000 de
nucleotide), ce formează împreună un haplotip; în populaţie, indivizi diferiţi
au haplotipuri diferite. Asocierea puternică între SNPs dintr-o regiune permite
ca, prin identificarea numai a câtorva SNPs (numite SNPs „etichetă“), alese cu
grijă – A/G, T/C şi C/G, din aceeaşi figură (c)
– să se reconstituie restul SNPs din haplotip. De exemplu, în figură (b), modelul A-T-C este caracteristic
haplotipului 1, C-A-A haplotipului 2 etc.
ProiectulHapMap s-a desfăşurat în trei faze,
diferite prin numărul de SNPs analizate, numărul de persoane şi populaţii
studiate şi tehnicile folosite. În fazele I (2005) şi II (2007) au fost
cartografiate 3,1 milioane de SNPs comune1(1 la 1.000 pb), la 270 de persoane (90 de „trios“ – doi părinţi şi un copil) din
patru populaţii din Europa, Africa şi Asia. Faza III (HapMap3), finalizată în septembrie 2010, a permis – prin
ameliorarea tehnologiilor utilizate – analiza altor 1,6 milioane SNPs (mai
rare) dar şi a 1.610 variaţii ale numărului
de copii (CNV), ce acoperă 12% din genom, la 1.184 de persoane, din 11
populaţii diferite. Am subliniat aceste detalii pentru a evidenţia rezultatele
deosebite ale acestui proiect, care fac din HapMap
un instrument performant pentru identificarea genelor de susceptibilitate la
boli comune. Pe baza cartografierii pe cromozomii umani a markerilor
polimorfici de tip SNPs, s-au dezvoltat tehnici de genotipare cu debit mare, de
tipul hibridării pe microreţele („microcipuri“) ADN, care, printr-o singură
„scanare“ a ADN al unei persoane, determină simultan tipul şi poziţia a circa
un milion de SNPs din întregul său genom.
În
ultimii cinci ani, s-a realizat un salt spectaculos al tehnologiilor de secvenţiere
a ADN, care au înregistrat o creştere progresivă a preciziei şi vitezei,
concomitent cu reducerea semnificativă a costurilor. A fost astfel posibilă
analiza genomului diploid (şase
miliarde de nucleotide) al unor persoane2,
iar primele rezultate (2007–2008), comparate cu secvenţa de referinţă (haploidă)
determinată prin PGU (2004), au evidenţiat marea
diversitate a variaţiilor structurale, dar mai ales faptul că procentul lor
este de patru-cinci ori mai mare decât
se stabilise iniţial (0,1% în schiţa genomului uman din 2001). În aceste condiţii,
s-a lansat, în 2008, proiectul „1.000 de
genomuri“, care îşi propune stabilirea unui catalog complet şi detaliat al
tuturor variantelor structurale, comune şi rare, în diferite grupuri etnice,
care să permită determinarea riscurilor de boală şi personalizarea
tratamentului.
După
catalogarea şi cartografierea celor mai frecvente variaţii structurale din
genomul uman şi introducerea tehnicilor de genotipare cu debit mare pe microreţele
(cipuri) ADN de tip SNPs, s-au iniţiat studii
de asociere la nivelul întregului genom („genome-wide
association studies“ – GWAS) între diferite variante structurale şi genele
de susceptibilitate la boli comune. Aceste studii se bazează pe ipoteza că
genele de susceptibilitate se asociază mai frecvent decât ne aşteptăm3 cu
anumiţi markeri genetici (SNPs) ce definesc o anumită regiune genomică sau
haplotip. Prin scanarea unui milion de SNPs din întregul genom la un grup de bolnavi şi la un grup control se poate stabili dacă un
haplotip (definit printr-un anumit marker, SNPs „etichetă“) are o frecvenţă
semnificativ mai mare la bolnavi comparativ cu martorii sănătoşi; în acest caz,
se poate presupune că în regiunea respectivă se află o genă implicată în
patogenia bolii. Identificarea „genei candidat“ (diferită de markerul SNPs ce
caracterizează regiunea) şi determinarea mecanismului prin care această genă
participă la producerea bolii necesită, evident, investigaţii ulterioare.
Până
în 2011, au fost efectuate peste 1.200 de studii de asociere la nivelul întregului
genom (GWAS), care au examinat peste 200 de boli comune, identificându-se peste
4.000 de SNPs asociate cu diferite boli multifactoriale. Aceste asocieri sunt
concordante cu ipoteza „boală comună –
variantă comună“, potrivit căreia influenţele genetice în susceptibilitatea
la boli comune sunt atribuite unui număr limitat de variante prezente la mai
mult de 1–5% din populaţie. Printre primele succese ale acestor studii se numără
identificarea unor gene asociate cu degenerescenţa maculară (CFH), boala
coronariană (CDKN2A/2B), dislipidemii (SORT1), poliartrita reumatoidă
(TRAF1–C5), diabetul zaharat (TCF7L2; SLC30A8; CDKAL1), astmul bronşic (ORMDL3;
GSDML), obezitatea (INSIG2, FTO) etc.
Identificarea
genelor de susceptibilitate deschide calea medicinii
predictive, ce va permite depistarea precoce a persoanelor cu risc crescut pentru anumite boli comune şi
iniţierea unor măsuri preventive
personalizate. Paradoxal, cu toate rezultatele obţinute, progresele făcute în
descifrarea rolului factorilor genetici în vulnerabilitatea (şi deci riscul) la
bolile complexe, multifactoriale sunt încă modeste. Explicaţiile posibile şi
perspectivele de viitor vor fi discutate într-un alt articol.
Descifrarea
mecanismelor patogenice în care sunt implicate genele de susceptibilitate va
permite noi abordări terapeutice. De
exemplu, studiile GWAS în degenerarea
maculară legată de vârstă (una din cauzele principale de tulburări ale
vederii centrale peste 50 de ani şi apoi orbire) au identificat la bolnavi două
SNPs situate în gena CHF pentru factorul complement H, care cresc răspunsul
inflamator (mediat de calea alternativă a sistemului complement) în maculă;
acest fapt surprinzător a generat cercetări promiţătoare privind identificarea
persoanelor cu risc crescut de a face degenerare maculară peste 50 de ani şi
dezvoltarea unor terapii inhibitorii ce implică sistemul complement.
Dacă
identificarea factorilor de risc individual la anumite boli comune defineşte
latura „predictivă şi preventivă“ a medicinii genomice, corelarea unor variante structurale specifice ale genomului uman cu
eficienţa unor intervenţii terapeutice deschide calea „medicinii
personalizate“, deoarece adaptează şi optimizează terapia la profilul genetic
al pacientului, ajustând-o la necesităţile sale. În articolul anterior
exemplificam această idee cu prezentarea variantelor genetice asociate cu răspunsul
diferit la tratament al bolnavilor în hepatita cronică cu virus C. Se pot da şi
alte exemple care demonstrează că medicaţia personalizată poate fi definită
prin sintagma „medicamentul potrivit
pentru pacientul potrivit, la momentul optim şi în doza adecvată“. Cercetările
de farmacogenomică efectuate până în prezent sunt promiţătoare, justificând un
optimism temperat în realizarea terapiei personalizate. Dar, despre toate
acestea vom discuta, de asemenea, în articolele viitoare.
Implementarea
clinică a medicinii genomice va necesita însă depăşirea altor obstacole. Cel
mai important este reprezentat de necesitatea
introducerii unor tehnologii de
secvenţiere a genomului personal, rapide şi la un cost acceptabil, pentru
ca determinarea profilul individual
al variantelor structurale genomice să fie accesibilă în practica curentă.
Progresele realizate în această direcţie sunt uluitoare: la 10 ianuarie 2012 (o
dată ce ar putea deveni „ziua genomului“),
două companii concurente (Ilumina Inc.
şi Life Technologies Corp.) au anunţat
realizarea unor nanomaşini de secvenţiere capabile „să citească“ genomul uman în
24 de ore, la preţul de 1.000 de dolari per genom, considerat de mult timp „o
piatră de hotar“ în genomică. Se apropie momentul în care secvenţierea completă
a genomului uman va deveni o metodă de rutină a arsenalului diagnostic. Aceasta
ridică însă noi şi delicate/dificile probleme pentru medici, dar şi pentru
sistemul de sănătate publică şi pacienţi:
• Cine
va beneficia de secvenţierea completă a genomului?
• Când
ar trebui făcută această secvenţiere în decursul vieţii individului?
• Cine
va interpreta informaţiile obţinute despre structura genomului pacienţilor,
pentru a stabili fie măsuri adecvate de prevenire a bolii la persoanele cu
risc, fie o schemă terapeutică individualizată (cu maximum de beneficii şi
minimum de efecte adverse) elaborată „în
timp real, la patul bolnavului“?
• Cum vor putea fi evitate eventualele
discriminări (la angajare, la asigurare) produse de „scurgerea“ de informaţii
genomice personale (de exemplu, riscul de boală)?
Cert este că
implementarea clinică a medicinii genomice este o problemă „de viitor
apropiat“, pentru care toţi clinicienii trebuie să fie „cognitiv pregătiţi“.