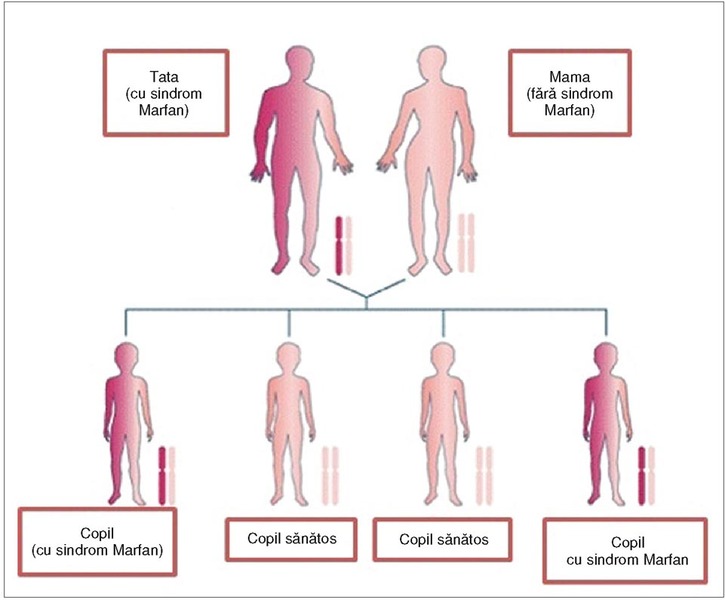
Sindromul Marfan este o boală genetică rară a ţesutului conjunctiv, ce
face parte din grupul fibrilinopatiilor, afecţiuni caracterizate printr-un
număr de semne clinice comune, care însă se asociază în mod diferit de la o
boală la alta. Este cauzat de o mutaţie în gena care determină producţia de
fibrilină 1, FBN1. Fibrilina este esenţială în formarea fibrelor elastice ale
ţesutului conjunctiv. Gena FBN1 este situată pe cromozomul 15 şi se transmite
după modelul autozomal dominant. Mutaţii ale FBN1 determină secreţie excesivă
de factor de creştere transformator beta (TGF-β). Creşterea TGF-β cauzează
probleme în ţesuturile conjunctive din tot corpul, ceea ce determină apariţia
caracteristicilor clinice asociate sindromului Marfan.
Boala
afectează aproximativ 1 din 5.000 de indivizi, bărbaţi şi femei, indiferent de
rasă sau grup etnic. Aproximativ trei sferturi din persoanele cu sindromul
Marfan l-au moştenit de la unul din părinţi. În cazul mutaţiilor spontane, se
naşte un copil cu sindrom Marfan fără a mai exista boala în familie. Există 50%
risc ca o persoană cu sindrom Marfan să transmită mutaţia specifică
descendenţilor săi.
Istoric. Numele
sindromului provine de la pediatrul francez Antoine-Bernard Marfan, care în
1896 a prezentat cazul unei fetiţe de 5 ani care avea o înfăţişare deosebită şi
prezenta câteva din caracteristicile clinice ale sindromului Marfan. În 1968,
Hugh Bentall a realizat o intervenţie chirurgicală care îi poartă numele, prin
care a înlocuit aorta ascendentă şi valva aortică la un pacient cu această
boală. Gena ale cărei mutaţii determină boala a fost identificată de Francesco
Ramirez la Mount Sinai Medical Center din New York, în 1991.
Medical Center din New York, în 1991.
Semne şi
simptome clinice. Deoarece ţesutul conjunctiv se găseşte în tot corpul,
sindromul Marfan are o afectare multisistemică. Cele mai frecvent afectate sunt
inima, vasele sanguine, oasele, articulaţiile şi ochii. Afectarea ţestului
pulmonar poate conduce la pneumotorax. Afectarea aortei poate determina
apariţia dilataţiei aortice sau formarea anevrismului. Pacienţii pot prezenta
cataractă şi subluxaţie de cristalin.
Manifestările clinice ale sindromului Marfan
sunt variabile, atât între membrii aceleiaşi familii (care prezintă aceeaşi
alelă mutantă), cât şi între indivizi din familii diferite (şi care pot
prezenta alele mutante diferite), fenomen numit expresivitate variabilă.
Criteriile de diagnostic au fost definite iniţial la Berlin în 1988 şi reactualizate
în 1996, la Gand.
Prezenţa
simultană a membrelor lungi, a subluxaţiei de cristalin şi a dilatării
rădăcinii aortice este, în general, suficientă pentru diagnostic. Există peste
30 de alte caracteristici clinice asociate cu acest sindrom, majoritatea implicând
scheletul, pielea şi articulaţiile. Alte semne clinice: pectus excavatum sau pectus
carinatum; picior plat; boltă ogivală, dinţi înghesuiţi; flexibilitate
articulară excesivă (cu excepţia coatelor); dizabilităţi de învăţare; dizlocare
a cristalinului, miopie; scolioză; faţă subţire şi îngustă.
Literatura
de specialitate descrie mai multe forme clinice de boală: forma clasică a
sindromului Marfan; forme incomplete de sindrom Marfan: • forma fără semne
oculare • forma cu degete lungi • forma neonatală şi infantilă, cu debut
precoce, mai gravă decât forma clasică, ce poate duce la pierderea mobilităţii
articulaţiilor şi la deces precoce prin insuficienţă cardiacă • ectopia
cristalinului • forme familiale de anevrism de aortă.
În
2010, un grup de experţi a revizuit criteriile de diagnostic pentru sindromul
Marfan. Acestea sunt:
* În absenţa
oricăror antecedente familiale de boală:
–
Anevrism de rădăcină aortică cu scor Z ≥ 2 pentru vârstă şi sex sau ectopia
(dezlipirea) de cristalin (ectopia
lentis). Prezenţa acestor anomalii stabileşte fără echivoc diagnosticul,
indiferent de asocierea altor semne sistemice specifice, cu excepţia cazurilor
când ele sunt orientative pentru sd. Shprintzen-Goldberg, sd. Loeys-Dietz sau
sd. Ehlers-Danlos.
– Anevrism de rădăcină aortică cu scor Z ≥ 2
pentru vârstă şi sex, cu sau fără disecţie, şi prezenţa mutaţiei FBN1;
– Anevrism de rădăcină aortică cu scor Z ≥ 2
pentru vârstă şi sex, şi scor sistemic ≥ 7;
– Ectopia lentis asociată cu mutaţie FBN1.
* În
prezenţa unui istoric familial de boală:
– Ectopia
lentis şi antecedente heredocolaterale de boală;
– Scor sistemic ≥7 şi antecedente
heredocolaterale de boală;
– Anevrism de rădăcină aortică cu scor Z ≥7
peste 20 de ani sau ≥3 sub 20 de ani, asociat cu istoric familial de boală.
Pentru
determinarea scorului sistemic, există programe specializate automate care îl
calculează în funcţie de simptomatologia clinică
(http://www.marfan.org/dx/score).
Diagnosticul
diferenţial
se face în funcţie de vârstă: sub 20 de ani, cu boli nespecifice ale ţesutului
conjunctiv, iar peste 20 de ani, cu sindromul MASS (miopie, prolaps de valvă
mitrală, dilataţie de rădăcină aortică, anevrism de aortă, vergeturi şi
tulburări scheletale), prolapsul  de valvă mitrală. De asemenea, mai trebuie diferenţiat
de sd. Loeys-Dietz, sd. Shprintzen-Goldberg, sd. Ehlers-Danlos, sd. Beals
(arahnodactilia congenitală contracturală), homocistinuria şi sd. Stickler.
de valvă mitrală. De asemenea, mai trebuie diferenţiat
de sd. Loeys-Dietz, sd. Shprintzen-Goldberg, sd. Ehlers-Danlos, sd. Beals
(arahnodactilia congenitală contracturală), homocistinuria şi sd. Stickler.
Prognosticul
bolii depinde în cea mai mare parte de gradul de afectare cardiacă. Cu toate
acestea, majoritatea pacienţilor au o speranţă de viaţă crescută.
Tratament. Nu există
tratament specific al bolii. Terapiile simptomatice, adjuvante, au rolul de a
îmbunătăţi calitatea vieţii acestor pacienţi şi de a creşte speranţa de viaţă.
Cel mai important lucru este monitorizarea stării de sănătate a aortei prin
consultaţii de specialitate, ecocardiografii şi alte tehnici de depistare a
modificărilor cardiovasculare. Dacă aorta este dilatată, este recomandat
tratamentul pentru scăderea tensiunii arteriale. De asemenea, se recomandă
evitarea eforturilor fizice intense care pot suprasolicita cordul. Uneori,
intervenţiile chirurgicale la nivelul aortei pot fi salvatoare de vieţi. În
cazul pacienţilor care prezintă deviaţii patologice ale coloanei vertebrale
poate fi necesară intervenţia chirurgicală şi/sau fizioterapia. Problemele
oculare trebuie tratate cât mai curând posibil. De asemenea, sunt recomandate
tratamentul şi monitorizarea anomaliilor coloanei vertebrale, în special în
adolescenţă. Femeile cu sindrom Marfan însărcinate necesită o supraveghere
suplimentară din cauza suprasolicitării cardiace din perioada sarcinii.
Complicaţiile includ progresia rapidă a dilatării aortice, disecţie şi ruptură
aortică în timpul sarcinii, naşterii sau în perioada postpartum.
Monitorizarea
şi tratamentul pacienţilor presupune colaborarea eficientă a unei echipe
multidisciplinare formată din medicul de familie, pediatru, cardiolog,
oftalmolog, genetician, chirurg ortoped, psiholog.
Complicaţii.Dintre
posibilele complicaţii asociate sindromului Marfan, cele mai de temut sunt cele
cardiace. Regurgitarea aortică, ruptura de valvă aortică, endocardita
bacteriană, anevrismul disecant de aortă, insuficienţa cardiacă, prolapsul de
valvă mitrală sunt cele mai frecvente afecţiuni ameninţătoare de viaţă.
 – bolnavul îşi poate înconjura încheietura mâinii cu degetele I şi II reunite, care chiar se depăşesc unul pe altul în timpul acestei acţiuni") Persoanele
cu sindrom Marfan trebuie să evite agenţii care stimulează sistemul
cardiovascular, sporturile ce presupun contactul fizic direct, sportul de
performanţă, exerciţiile de tip izometric şi să practice cu moderaţie exerciţii
de tip aerobic.
Persoanele
cu sindrom Marfan trebuie să evite agenţii care stimulează sistemul
cardiovascular, sporturile ce presupun contactul fizic direct, sportul de
performanţă, exerciţiile de tip izometric şi să practice cu moderaţie exerciţii
de tip aerobic.Dacă
afectarea cardiacă este diagnosticată precoce, corect monitorizată şi
controlată terapeutic, aceşti pacienţi pot avea o viaţă cotidiană obişnuită.
Sfat
genetic. 75%
din pacienţi au un părinte afectat şi în 25% din cazuri afecţiunea apare
secundar unei mutaţii de novo în ovul
sau în spermatozoid, ambii părinţi fiind sănătoşi. Un individ afectat are un
risc de 50% de a avea copii afectaţi, indiferent de sexul acestora. Dacă
părinţii sunt sănătoşi şi au un copil afectat, riscul de a mai avea un alt
copil afectat este mai mic de 50%, dar mai mare decât cel din populaţia
generală, considerând posibilitatea unui mozaicism germinal (mai multe celule
germinale afectate de mutaţie).
Testarea
prenatală pentru sindromul Marfan, precum şi diagnosticul genetic
preimplantator sunt posibile, dar asemenea cereri sunt foarte rare considerând
faptul că această afecţiune nu este caracterizată de deficienţe intelectuale şi
că astăzi există posibilităţi terapeutice care permit o durată de viaţă normală
şi o calitate a vieţii apropiată de cea a persoanelor normale.
De
altfel, percepţia publică a sindromului Marfan este una foarte bună, fiind
cunoscuţi sportivi olimpici (Flo Hyman), dar şi muzicieni şi compozitori
(Jonathan Larson, Niccolò Paganini, Serghei Rahmaninov, Robert Johnson şi Sir
John Tavener) care au avut sau este posibil că au avut sindrom Marfan.
Grupuri de
sprijin. La
nivel naţional, în cadrul Centrului de boli rare NoRo din Zalău, se oferă
sprijin psihomedical atât pacienţilor, cât şi familiilor acestora, prin
întâlniri de grup. Pacienţi din toată ţara sunt monitorizaţi şi instruiţi
pentru a-şi controla cât mai corect boala. La nivel internaţional, Fundaţia
Marfan, www.marfan.org, este o platformă web care oferă sprijin şi informare
pentru pacienţii cu această boală şi familiile lor în lupta împotriva bolii şi
a tulburărilor conexe.