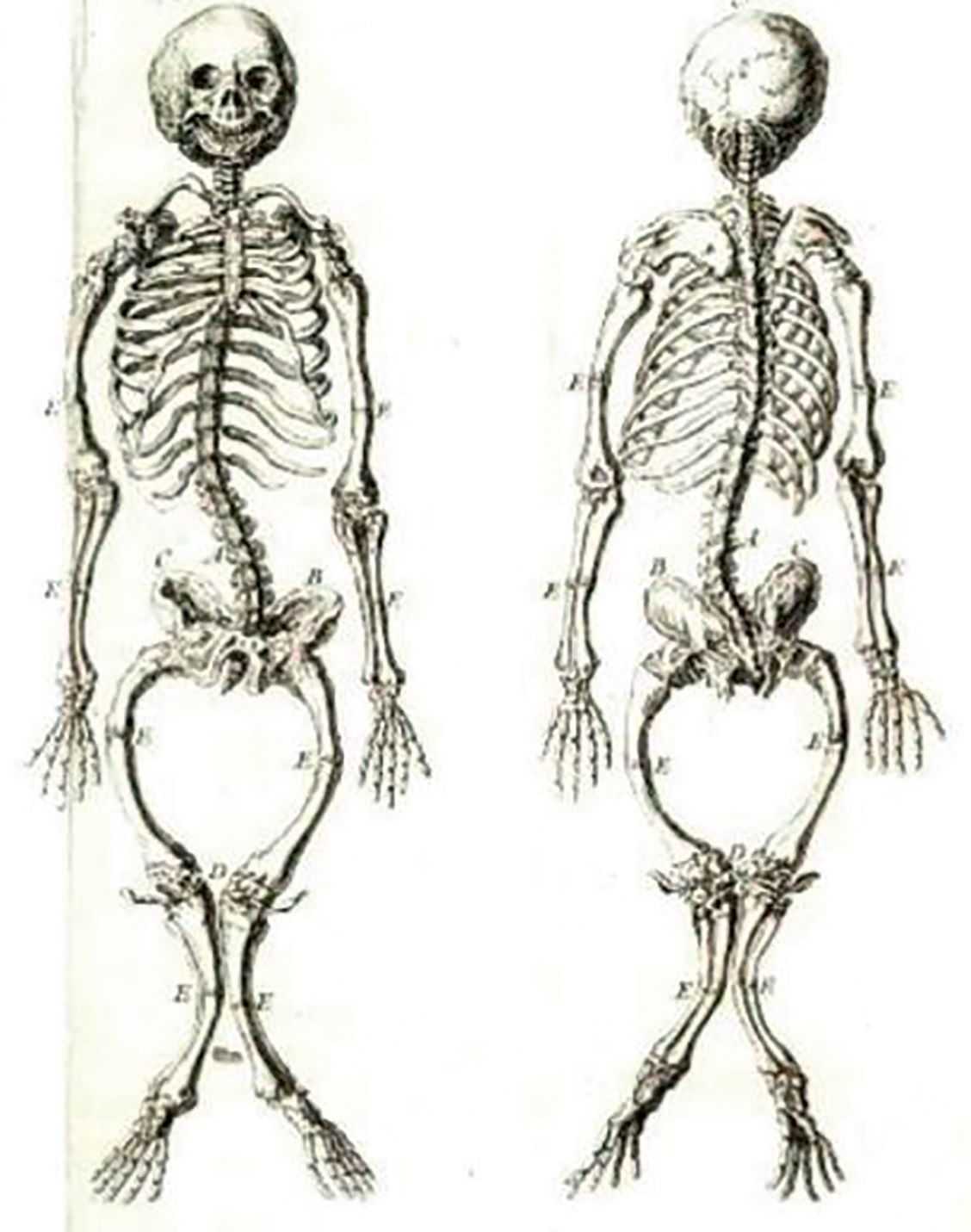
Rahitismele genetice sunt maladii generate de tulburări ale metabolismului fosfocalcic.
Concentraţia sărurilor fosfocalcice este influenţată de mai mulţi factori, care depind de glandele endocrine, de tubul digestiv, ficat și rinichi. Fiziopatologia acestora poate genera tulburări funcţionale, ca urmare a unor boli genetice care interesează aceste aparate și organe. Rahitismele genetice sunt forme de rahitism rezistente în grad variabil la tratamentul cu vitamina D, motiv pentru care se mai numesc și rahitisme vitamino-D-rezistente.
Este o formă rară de rahitism. Incidenţa este de 1 la 100.000 de nou-născuţi vii. Tulburarea metabolismului fosfocalcic este caracterizată clinic printr-un defect de mineralizare osoasă și biochimic prin activitatea deficitară a fosfatazei alcaline. Cea mai mare parte a fosfatazei alcaline este produsă la nivelul ficatului și oaselor, iar cantităţi mai mici sunt produse în intestin, rinichi și placentă.
Descrisă pentru prima dată de Rathbun în 1948 la un nou-născut de 9 săptămâni, hipofosfatazia a fost ulterior studiată de mai mulţi autori, care au adus contribuţii importante mai ales în domeniul genetic. În 1957, Fraser clasifica rahitismul hipofosfatazic, în raport cu vârsta de debut, în mai multe forme: perinatală, infantilă sau neonatală, a copilăriei și adultă.
Biologie
Fosfataza alcalină nonspecifică, TNSALP (tissue non-specific alkaline phosphatase), deficitară la nivelul osteoblastelor și condrocitelor, perturbă mineralizarea osoasă, inducând rahitism și osteomalacie. Valorile scăzute ale TNSALP reprezintă un semn patognomonic și sunt determinate de una dintre cele 200 de mutaţii genetice care pot surveni la nivelul genei ALPL, ce codifică TNSALP. Transmiterea genetică se face autosomal recesiv pentru formele perinatale, infantile sau neonatale, iar pentru celelalte forme, fie autosomal recesiv, fie autosomal dominant.
Fiziopatogenic, TNSALP produce hidroliza câtorva substanţe, printre care pirofosfatul anorganic și piridoxal 5-fosfat, o formă importantă a vitaminei B6. Pirofosfatul anorganic se acumulează în afara celulei, când TNSALP are valori scăzute, și inhibă formarea de hidroxiapatită, una dintre principalele componente osoase, determinând rahitism la nou-născuţi și copii și osteomalacie la adulţi. Piridoxal 5-fosfat este principala formă de vitamina B6 și trebuie defosforilată de TNSALP pentru a putea traversa membrana celulară. Deficienţa vitaminei B6 la nivelul creierului produce convulsii.
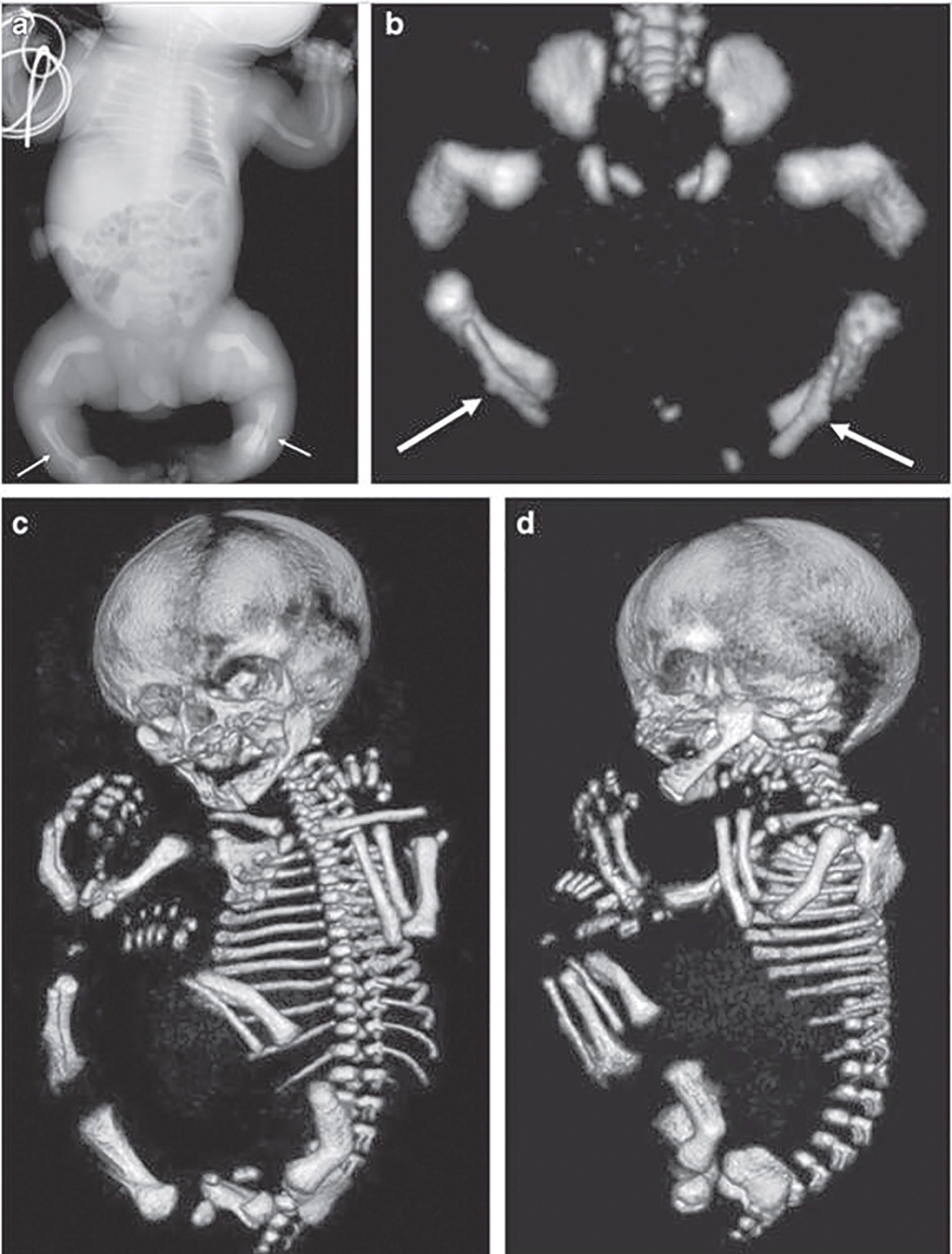
Rahitismul hipofosfatemic perinatal este forma cu cea mai mare mortalitate. În perioada fetală și la naștere, membrele sunt mai scurte și deformate, coastele deformate și distorsionate, iar craniul – intens demineralizat. Această formă produce rapid moartea prin tulburări respiratorii. Numărul nou-născuţilor morţi nu se cunoaște, iar supravieţuirea pe o durată îndelungată este rară. Nou-născuţii care supravieţuiesc prezintă tulburări respiratorii, osteomalacie și plămâni subdezvoltaţi, hipoplazici. Insuficienţa respiratorie poate fi cauză tardivă de deces. Epilepsia poate surveni și adesea este letală. Nemineralizarea osoasă extinsă determină anemie mieloplastică. Semnele caracteristice, simptomele și complicaţiile hipofosfataziei perinatale se pot grupa în manifestări scheletale (diformităţi toracice, încurbări, craniostenoză) și tulburări respiratorii.
Indisponibilitatea terapiei de implantare a enzimei deficiente face ca rata mortalităţii la acest segment de pacienţi să fie între 58% și 100%. În formele mai puţin agresive apar numai încurbarea oaselor lungi și pintenul Bowdler (vezi fig).
Sunt unii autori care susţin că rata mortalităţii este de 100%.
Incidenţa în Canada în 1957 a fost de 1 la 100.000 de nou-născuţi vii. Însă, fiind o afecţiune rară, adevărata incidenţă nu se cunoaște. Conform unei estimări din perioada 2000-2009, având la bază diagnosticul molecular, incidenţa acestei boli în Europa este de 1 la 538.000. O incidenţă crescută a fost întâlnită la comunităţile mennonite din Canada și în satele cu căsătorii endogame din Ungaria.
Formele care debutează în perioada copilăriei, de regulă după vârsta de 6 luni, au gravitate medie și prezintă hipotrofie staturoponderală și deformări ale membrelor, secundare deficitului de mineralizare osoasă și uneori consolidărilor vicioase ale unor fracturi.
La examenul clinic putem constata brăţări epifizare, mătănii costale sau căderea prematură a dinţilor. Fontanela anterioară poate rămâne deschisă o perioadă mai îndelungată.
Forma adultă, numită și odontofosfatazie, se manifestă numai prin căderea precoce a dinţilor. În unele cazuri se asociază și nanismul sau o talie scundă, fragilitate osoasă sau deviaţie axială a membrelor.
La examinările de laborator, fosfataza alcalină este scăzută constant, iar calcemia și fosfatemia sunt în limite normale sau crescute. În urină apare o cantitate crescută de fosfoetanolamină.
Imagistică
La examenul ecografic al fătului se evidenţiază pinteni osoși, semn patognomonic pentru rahitismul hipofosfatazic. Evaluarea osificării la fetuși se face prin studiu ecografic. Datele obţinute prin examenul radiologic al unor avortoni arată diferenţieri ale osificării în funcţie de vârsta acestora. La 11 săptămâni este absentă osificarea calotei craniene, a corpilor vertebrali cervicali, toracali, a sacrului, a ischionului și pubisului.
La 14 săptămâni apare osificarea tuturor corpilor vertebrali, însă rămâne absentă osificarea calotei craniene, a bazei craniului, ischionului și pubisului. La 15 săptămâni apare și osificarea ischionului și pubisului. Examenul radiologic, în forma infantilă sau neonatală, prezintă segmente de membru în care scheletul este vizibil parţial. Metafizele sunt lăţite și concave. Diafizele sunt distorsionate și cu zone de resorbţie chistică la nivelul metafizelor. Coastele sunt deformate și foarte subţiri. La copiii peste 6 luni metafizele sunt largi și scobite, iar suprametafizar apar zone de osteoliză care pretează la confuzia cu un proces osteomielitic, însă lipsește reacţia periostală. Nucleii de osificare apar tardiv, sunt hipomineralizaţi și de dimensiuni mai mici. Încurbările metafizare au grade variate de amplitudine.
Structura histologică este atipică. Ţesutul osos este slab reprezentat și apar insule de ţesut osteoid și cartilaginos.
Cartilajele de creștere au o structură deficitară în care lipsesc sărurile de calciu și coloanele cartilajului seriat.
Rahitismul de tip Royer are caracter familial evidenţiat de mulţi autori. Aceste date arată că maladia este, cu certitudine, genetică. Royer a descris această afecţiune ca hipercalciurie idiopatică. Ea prezintă manifestări de tip rahitic când este însoţită de hipocalcemie.
Excreţia excesivă de calciu are o serie de consecinţe, dintre care cea mai frecventă este boala litiazică renală.
Diagnosticul se stabilește pe baza datelor clinice, a investigaţiilor de laborator și imaginilor radiologice.
Studiile moleculare vor preciza localizarea genei reponsabile de acest tip de rahitism. Hipercalciuria secundară unui proces patologic cunoscut nu este inclusă în această formă de rahitism.
Boala debutează în primii trei ani de viaţă, cu accese de tetanie ca urmare a hipocalcemiei. Pierderea excesivă de calciu determină tulburări de creștere. Nanismul din hiercalciuria familială este însoţit de osteoporoză și de genu var sau valg. Aspectul radiologic al scheletului ilustrează imagini ale oaselor lungi cu corticală subţiată. Nucleii epifizari apar mai târziu și au volum concordant cu talia nanică.
Tratamentul ortopedic vizează modificările axiale și corectarea modificărilor în var sau în valg ale șoldurilor, genunchilor și gleznelor.
Tratamentul medicamentos cu vitamina D3 și calciu este ineficient în rahitismul genetic, motiv pentru care aceste rahitisme se mai numesc și rahitisme vitamino-D-rezistente. Ameliorările obţinute în unele cazuri survin ca urmare a tipurilor de rahitism și pe măsura maturizării scheletului.
Tratamentul ortopedic se aplică atunci când acești copii prezintă fracturi și în special la nou-născuţi, sugari și copiii mici.
Tratamentul chirurgical este în prezent de bază și vizează tratamentul complicaţiilor evolutive ce apar în rahitismele genetice: deviaţiile axiale, nanismul, fracturile spontane etc.
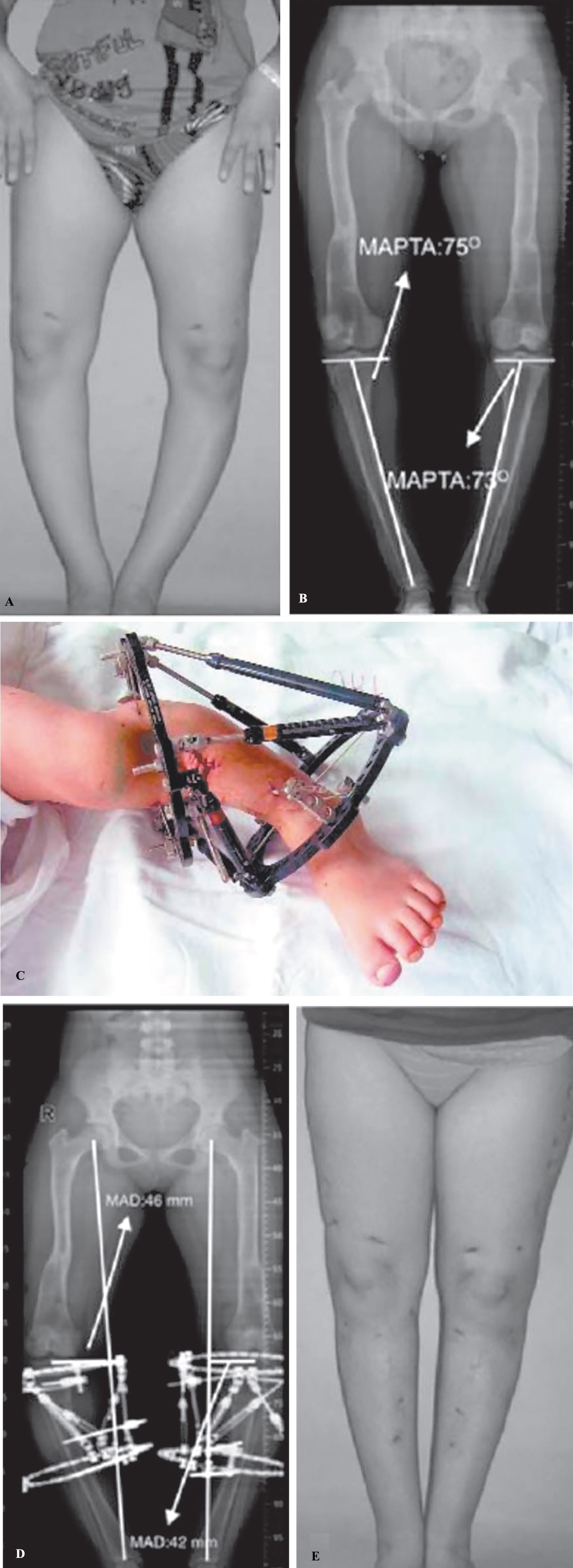
Deviaţiile axiale se evaluează periodic, prin studiul topogramei de bazin și de membre pelvine. Deviaţiile axiale nu sunt întotdeauna vizibile în ortostatism și în mers. Sunt situaţii în care membrele par normale, dar în realitate există un var sau un valg ce poate avea consecinţe mai târziu.
La copii, aceste diformităţi se tratează prin hemiepifiziodeză cu plăci în opt sau cu un șurub trecut transhemiepifizar. La adolescenţi se practică osteotomii subtrohanteriene, supracondiliene de femur sau proximale de tibie.
Încurbările diafizare sunt factori favorizanţi pentru apariţia fracturilor recurente. Locul amplitudinii maxime a încurbărilor se stabilește prin studiul axelor anatomice. Încurbările cu amplitudine mare impun uneori osteotomie dublă pentru a fi corectate.
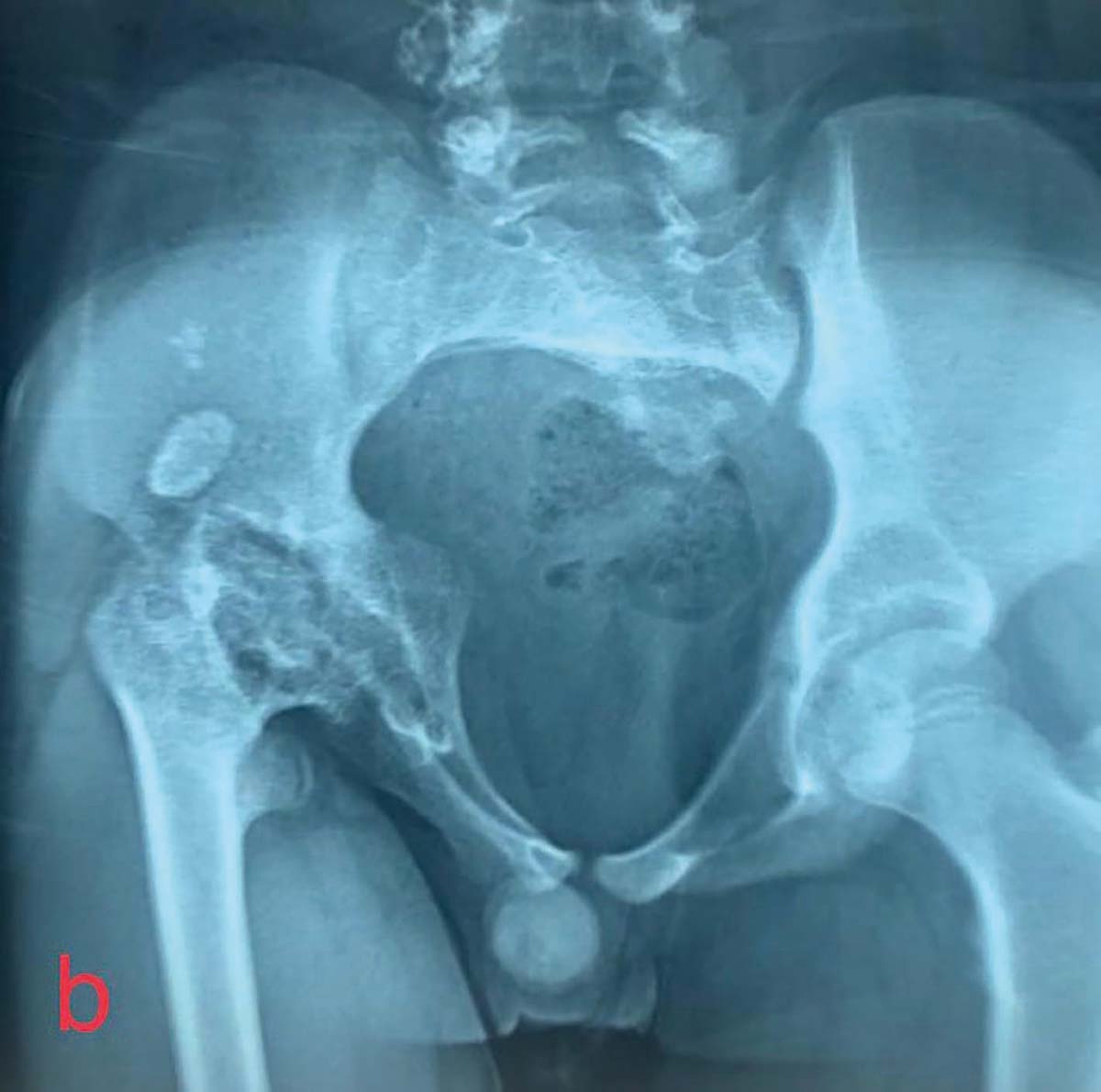
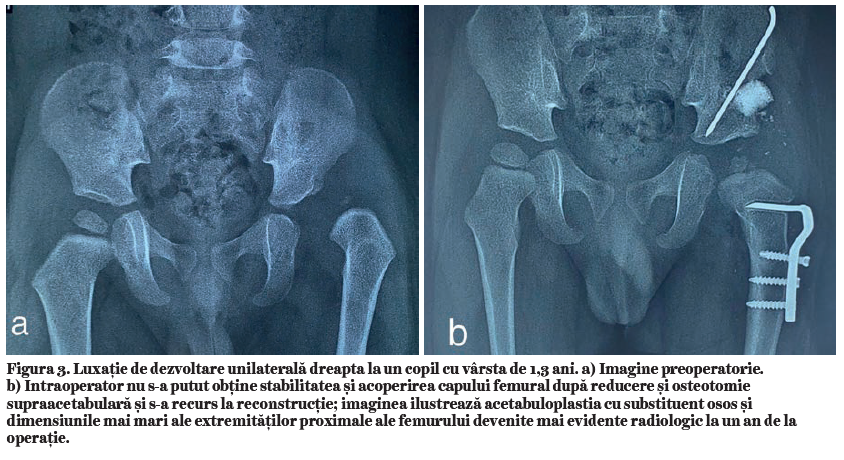
Când încurbările sunt însoţite de osteoporoză, se folosesc metode de osteosinteză complementară, plăci de reconstrucţie sau plăci pentru sinteza periprotetică (fig. 1).
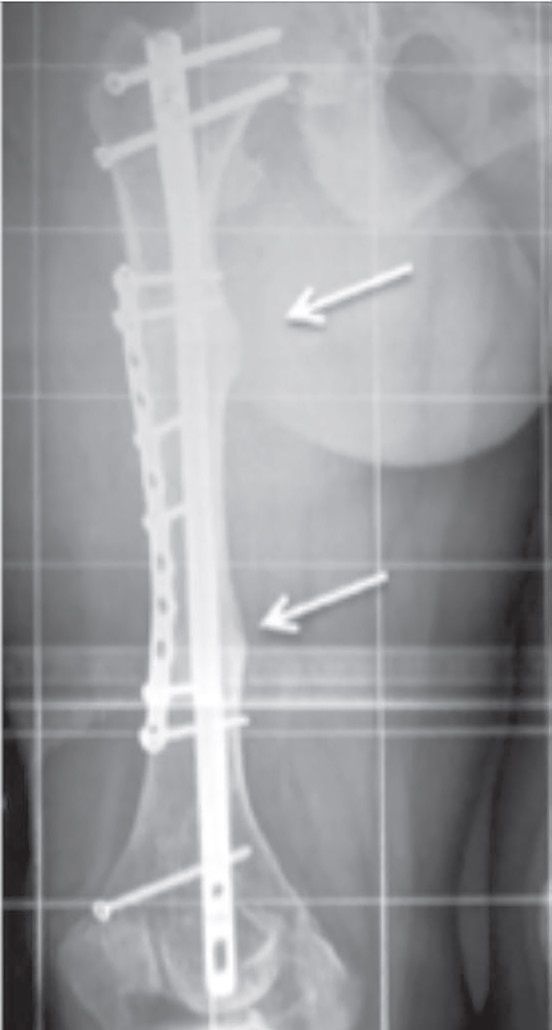
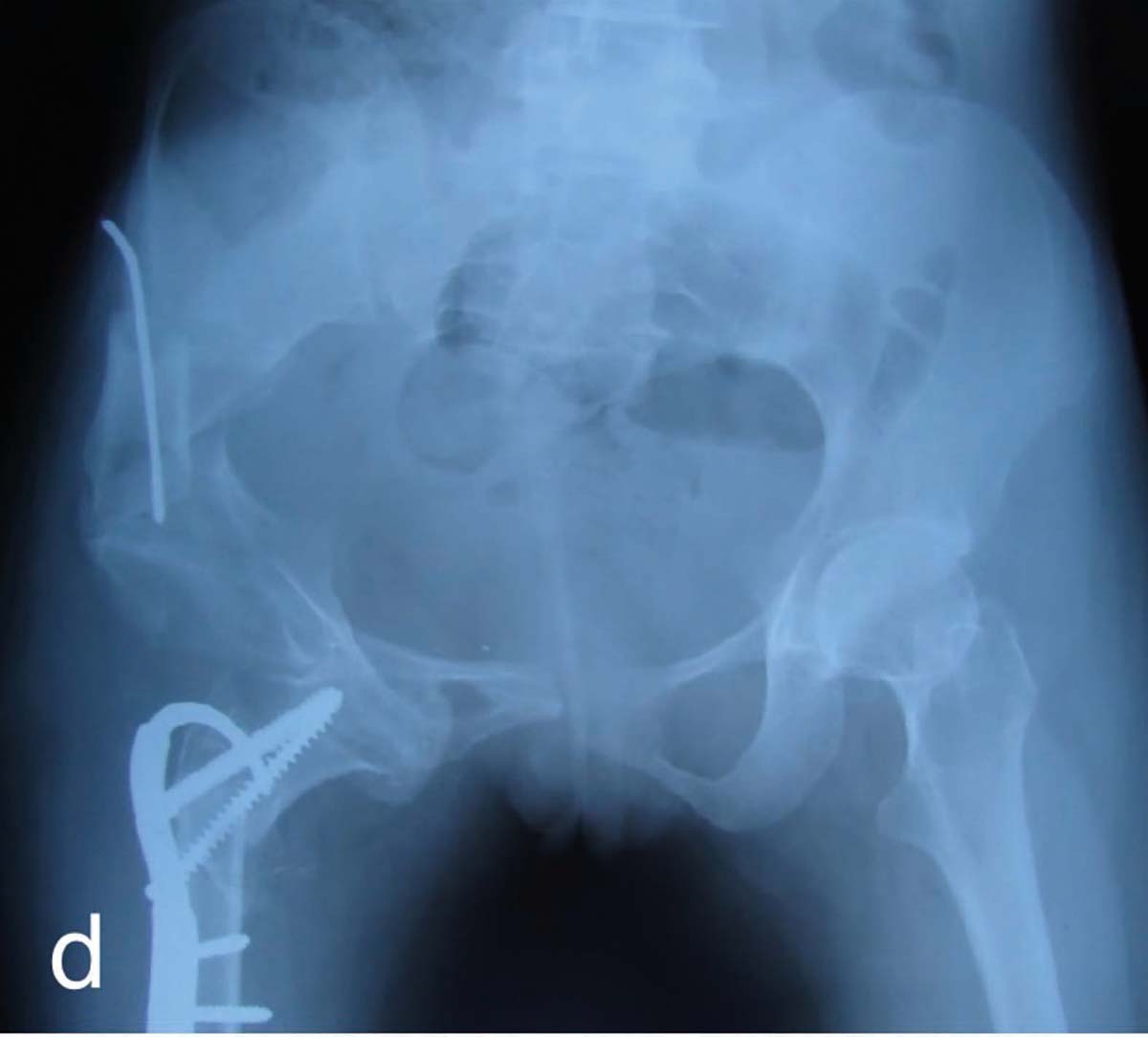
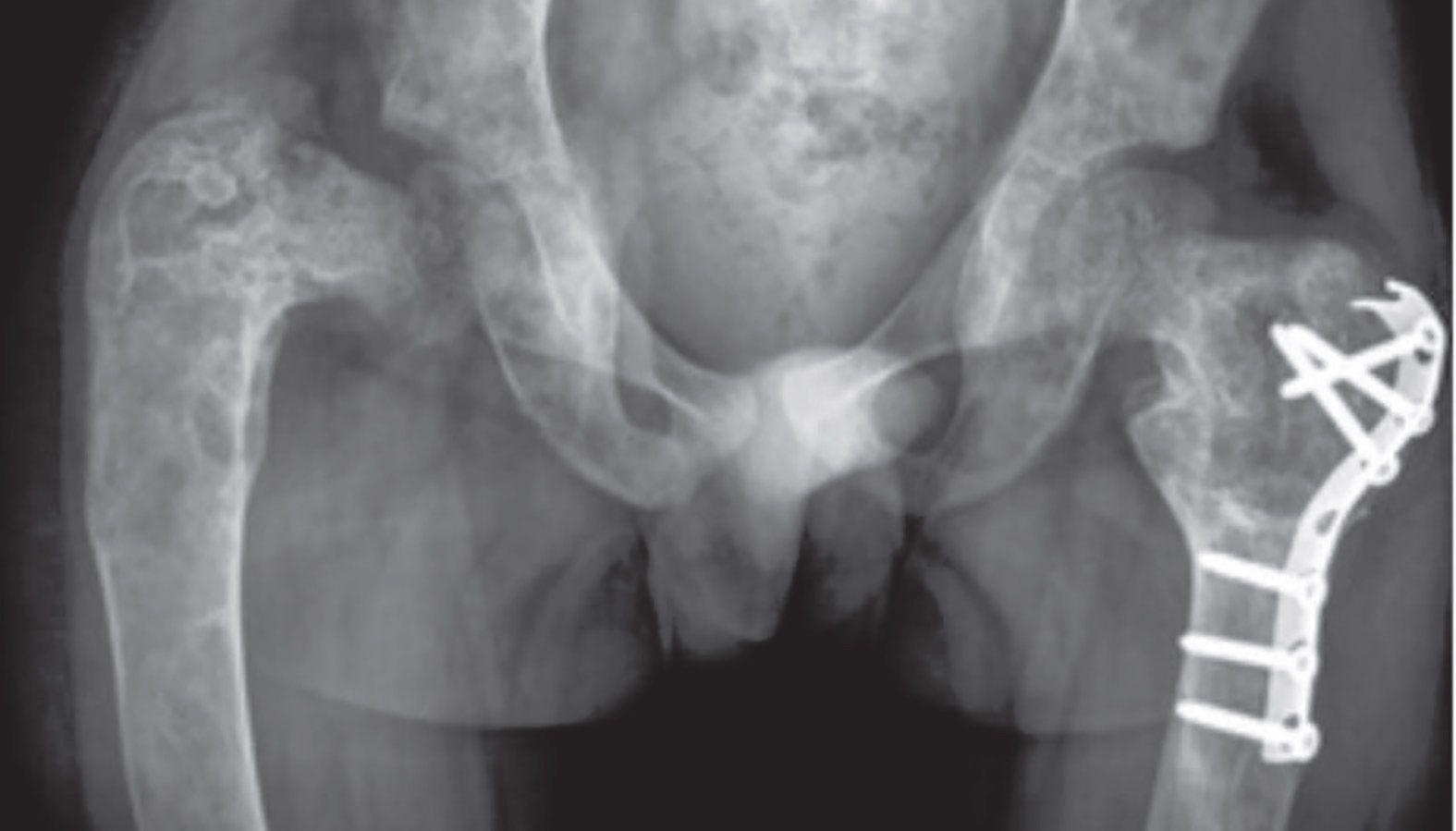
Nanismul apare la 40% dintre pacienţii cu rahitism genetic. Alungirile la nanicii rahitici se fac după principiile clasice, care constau în alungirea oaselor lungi cu o treime din lungimea lor și corectarea leziunilor asociate, sau după cele moderne: alungirea în dublu focar pe splint format din tije TEN, osteotomii oblice și corectarea concomitentă a diformităţilor în valg, var sau a încurbărilor, cu protecţia articulaţiilor. Fixatoarele de tip Taylor (fig. 2) sunt mai utile când apare slăbirea rezistenţei osoase, mai sigure și mai eficiente.
Fracturile spontane se pot trata cu tije TEN, cu plăci obișnuite, plăci de tip NBC, șuruburi sau broșe. Eficacitatea șuruburilor se asigură complementar și e greu de înţeles de începători sau de cei care nu cunosc tehnica osteosintezei sau care practică ortopedia pediatrică în mod teoretic. Artrozele apar uneori precoce, prin suprasolicitarea articulaţiilor incongruente, ca urmare a deviaţiilor axiale.
Protezele de șold și de genunchi necesită cunoștinţe și experienţa adaptată fiecărui caz în parte.
Dacă vrei să fii la curent cu tot ce se întâmplă în lumea medicală, abonează-te la „Viața Medicală”, publicația profesională, socială și culturală a profesioniștilor în Sănătate din România!
Titularii abonamentelor pe 12 luni sunt creditați astfel de:

Dacă vrei să fii la curent cu tot ce se întâmplă în lumea medicală, abonează-te la „Viața Medicală”, publicația profesională, socială și culturală a profesioniștilor în Sănătate din România!
Află mai multe informații despre oferta de abonare.
Cookie-urile ne ajută să vă îmbunătățim experiența pe site-ul nostru. Prin continuarea navigării pe site-ul www.viata-medicala.ro, veți accepta implicit folosirea de cookie-uri pe parcursul vizitei dumneavoastră.
Da, sunt de acord Aflați mai multe