Afecţiunile metabolic-ereditare hepatice sunt boli ce se manifestă încă din perioada neonatală sau din copilărie și sunt rezultatul unor deficienţe enzimatice hepatice condiţionate genetic.

Cel mai frecvent întâlnite sunt deficitul de alfa-1-antitripsină, boala Wilson și hemocromatoza ereditară.

UMF „Carol Davila”, București,
Medic primar Gastroenterologie,
Centrul de Gastroenterologie și Hepatologie,
Institutul Clinic Fundeni, București
Aceste afecţiuni se pot manifesta acut în perioada neonatală, evoluând ca o manifestare ameninţătoare de viaţă sau cu tablou de boală hepatică cronică, cu debut în copilărie, adolescenţă sau chiar la vârsta adultă, și progresie către ciroză hepatică și hepatocarcinom. Transplantul hepatic este de multe ori singura procedură salvatoare de viaţă în aceste afecţiuni. Studiile efectuate au stabilit faptul că aproximativ 5% din indicaţia de transplant hepatic în SUA este pentru boli metabolic ereditare hepatice pentru seriile de adulţi și până la 20% pentru seriile pediatrice (1).
Mai ales în populaţia pediatrică, aceste afecţiuni implică manifestări clinice polimorfe ce conduc la un important diagnostic diferenţial, mai ales cu intoxicaţiile acute sau neuroinfecţiile. Suspiciunea de boală hepatică metabolică se ridică la toţi copiii care prezintă sindrom de colestază, afectare progresivă neuromusculară, deficit de creștere. Creșterea aminotransferazelor, hepatomegalia, acidoza, hipoglicemia, hiperamoniemia, sindromul ascitic, coma trebuie să conducă la investigarea unei posibile boli metabolic-ereditare hepatice. În copilărie și adolescenţă, tabloul clinic îmbracă cel mai frecvent aspectul bolii hepatice cronice.
Anamneza familială cu prezenţa consangvinităţii, a avorturilor spontane multiple, a deceselor la vârstă tânără poate sugera antecedente heredocolaterale de boli hepatice metabolice.
Boala Wilson este o afecţiune transmisă autozomal recesiv ce se manifestă prin excreţie biliară deficitară a cuprului, cu acumularea sa la nivel hepatic și cerebral, consecinţă a unei anomalii genetice a proteinei ATP7B situată pe cromozomul 13. Această genă codifică o ATP-ază localizată la nivelul reţelei transgolgiene responsabilă de transportul intracelular al cuprului, atât cu încorporarea sa în apo-ceruloplasmină pentru sinteza ceruloplasminei, cât și cu excreţia sa biliară (5). Au fost descrise peste 500 de mutaţii la nivelul genei ATP7B, dintre care peste 380 de mutaţii sunt cauzatoare de boală. Frecvenţa mutaţiilor în populaţia generală este foarte crescută, de până la 1 la 90, cu o incidenţă medie a bolii de 1 la 30.000, dar cu largă variabilitate regională (2).
Boala poate fi diagnosticată la orice vârstă, cel mai adesea debutul fiind între 5 și 35 de ani, iar manifestările clinice sunt expresia afectării hepatice și neurologice. Cel mai vârstnic pacient diagnosticat cu boala Wilson a avut 80 ani, însă numai aproximativ 3% dintre cazuri sunt diagnosticate după decada a patra de viaţă.
Inelul Keiser-Fleischer, cauzat de depunerea patologică de cupru la nivelul membranei Descemet și evidenţiat în urma examenului oftalmologic cu lampa cu fantă, este întâlnit la peste 95% dintre pacienţii cu afectare neurologică, dar poate lipsi la pacienţii cu fenotip exclusiv hepatic, mai ales în populaţia pediatrică (3).
Semnele clinice de boală hepatică sunt nespecifice, iar afectarea hepatică poate precede manifestările neurologice cu până la zece ani. Gradul de afectare hepatică este variabil, de la hepatocitoliză izolată sau alterarea probelor funcţionale hepatice și până la ciroza hepatică. Principalele simptome și manifestări clinice întâlnite la pacienţii cu afectare hepatică în contextul bolii Wilson sunt: icterul, anorexia, vărsăturile (14-44%), hepato-splenomegalia (15-49%), hemoliza (5-20%), sindromul ascitic, edemele (14-50%), hemoragia digestivă superioară (3-10%), diateza hemoragipară (3-8%), insuficienţa hepatică acută (până la 17%).
Semnele neurologice cel mai frecvent întâlnite sunt reprezentate de tremor, ataxie, distonie. Manifestările clinice îmbracă un spectru polimorf, de la tulburări neurologice extrem de subtile și până la dizabilitate severă, cu sindrom akinetic-rigid, ataxie, sindrom distonic cu contracturi severe. În stadiile avansate de boală, pacientul este de obicei conștient, dar nu poate vorbi și nu se poate îngriji singur, din cauza incapacităţii de a-și controla mișcările și a distoniei severe.
Tulburările comportamentale și manifestările psihiatrice sunt, de asemenea, frecvent întâlnite și, de obicei, preced manifestările neurologice și hepatice. Ele variază de la scăderea capacităţii de atenţie și a performanţelor școlare până la tulburări de personalitate, impulsivitate, labilitate psihoemoţională, comportament inadecvat, exhibiţionism. De multe ori sunt confundate cu manifestările specifice pubertăţii. Manifestările psihotice severe, precum paranoia, schizofrenia, depresia, catatonia, sunt, de asemenea, întâlnite.
Manifestări mai rar întâlnite includ gigantismul, anomaliile renale (aminoaciurie, nefrolitiază, hipercalciurie, nefrocalcinoză), cardiomiopatie, miopatie, condrocalcinoză, osteoartrită, hipoparatiroidism, pancreatită, infertilitate, avorturi spontane repetate. Până la 23% dintre cazuri sunt complet asimptomatice și diagnosticate în contextul screeningului familial.
Pentru stabilirea diagnosticului este necesară o combinaţie de teste care să reflecte alterarea metabolismului cuprului. Atunci când inelul Keiser-Fleischer este prezent și ceruloplasmina are valori sub 0,1 g/l, diagnosticarea este facilă. Atunci când lipsește inelul Keiser-Fleischer (în cca. 50% dintre formele hepatice de boală), diagnosticul are la bază criteriile stabilite în 2001 la Leipzig de Grupul Internaţional de Lucru pentru boala Wilson (Tabelul) (4).
Acesta implică hepatita acută, cronică și ciroza hepatică de etiologii multiple. Boala Wilson cu debut fulminant trebuie suspectată în cazul unei hepatite acute cu icter și hemoliză. Manifestările neuropsihiatrice implică un diagnostic diferenţial complex cu alte tulburări comportamentale, iar IRM cerebrală este un demers diagnostic important.
Testarea genetică se recomandă în algoritmul de diagnostic, atunci când scorul Leipzig este ≤3 puncte. Majoritatea pacienţilor prezintă un genotip heterozigot compus. Mutaţia genetică cel mai frecvent întâlnită este polimorfismul de bază azotată unică H1069Q, din exonul 14 al genei ATP7B. La populaţia din România, mutaţia H1069Q a fost detectată în stadiul de homozigot la 21,1% dintre pacienţii cu boala Wilson, iar în stadiul de heterozigot compus, la 34,2%, cu o frecvenţă alelică de 38,1% (5).
Identificarea unei mutaţii la pacientul index are valoare pentru screeningul familial. Testarea prezenţei tuturor mutaţiilor descrise este dificil de implementat în practica de zi cu zi, dar devine posibilă odată cu dezvoltarea noilor metode de secvenţiere genică (Next genereration sequencing). Analiza de haplotip, care are la bază tiparul repetitiv di- și trinucleotidic ce flanchează gena ATP7B, este importantă pentru screeningul familial.
Principalele medicamente utilizate pentru terapia bolii Wilson sunt chelatorii de cupru și includ: D-penicilamina, trientina, zincul, tetratiomolibdatul și dimercaprolul. Ghiduri de practică pentru boala Wilson au fost publicate de EASL și de AASLD (6).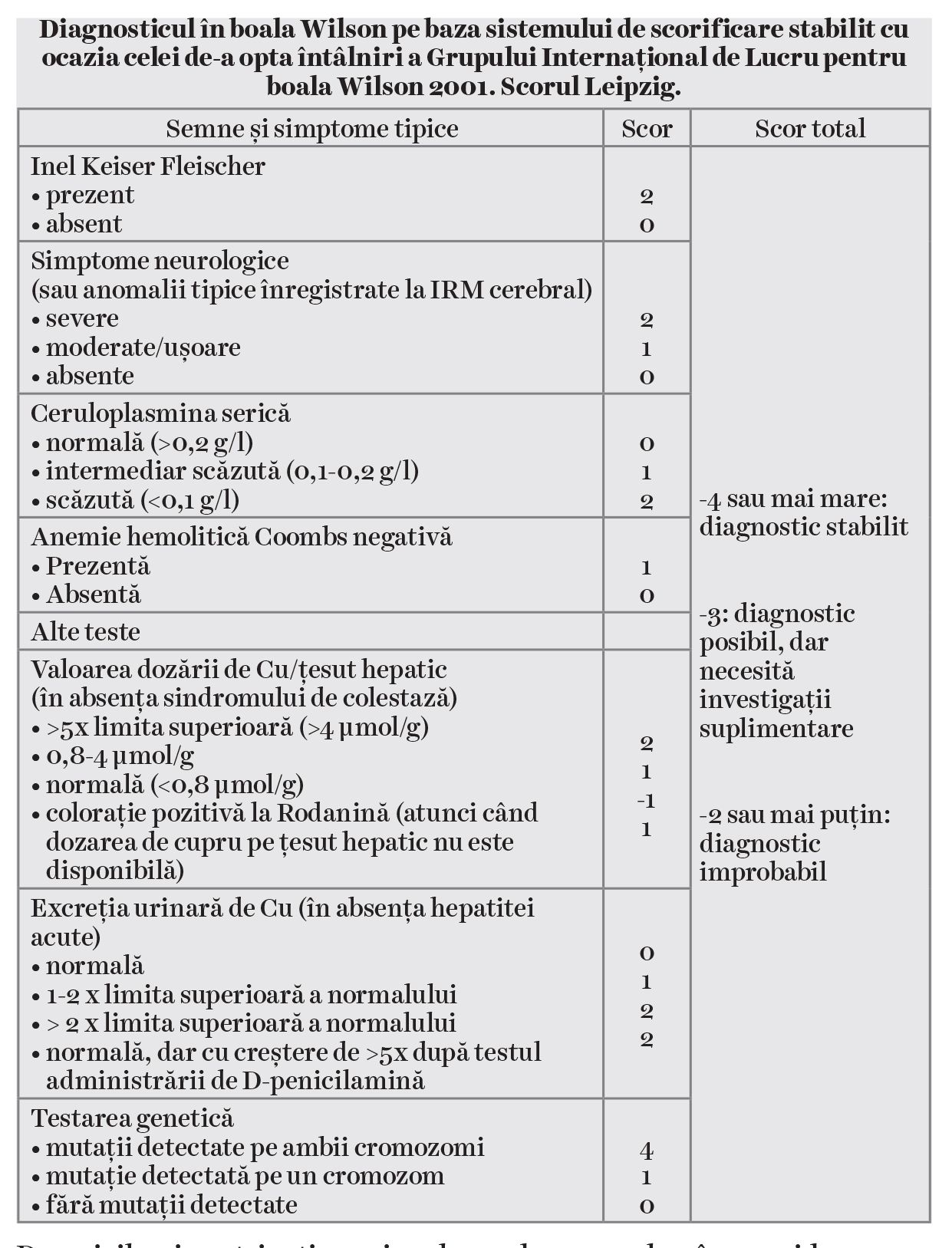 Ghidul EASL face următoarele recomandări terapeutice pentru managementul bolii Wilson:
Ghidul EASL face următoarele recomandări terapeutice pentru managementul bolii Wilson:
• Terapia iniţială pentru pacienţii simptomatici cu boală Wilson trebuie să includă un chelator de cupru (D-penicilamină sau trientina), trientina fiind mai bine tolerată.
• Zincul poate avea un rol ca terapie de primă linie la pacienţii cu afectare neurologică.
• Tratamentul pacienţilor presimptomatici sau al celor cu afectare neurologică în curs de tratament de menţinere se poate face cu chelator de cupru sau cu zinc.
• Tratamentul durează toată viaţa și nu trebuie întrerupt decât în cazul transplantului hepatic.
• Atunci când se utilizează tratamentul cu zinc, trebuie monitorizate periodic valorile aminotransferazelor și este recomandat tratament chelator dacă se înregistrează creșterea acestora.
• Pacienţii trebuie să evite consumul de alimente și apă cu conţinut crescut de cupru, mai ales pe durata primului an de terapie.
• În cazul insuficienţei hepatice acute cauzată de boala Wilson se va lua în considerare transplantul hepatic atunci când scorul King este ≥11 puncte.
• Pacienţii cu ciroză hepatică decompensată, nonresponsivi la terapie cu chelator de cupru, trebuie prompt evaluaţi pentru transplant hepatic.
• Tratamentul bolii Wilson trebuie continuat pe durata sarcinii, dar se va lua în considerare reducerea dozelor de trientină sau D-penicilamină.
• Pentru monitorizarea de rutină, se recomandă bianual examenul clinic neurologic și fizic, determinarea cupremiei, a ceruloplasminei serice, a enzimelor hepatice, INR, a hemoleucogramei și analiza urinei.
• Determinarea cantitativă urinară a cuprului/24 h trebuie efectuată anual în cursul terapiei și după două zile de oprire a terapiei.
• Estimarea valorii cuprului nelegat de ceruloplasmină este un parametru util pentru monitorizarea eficienţei terapiei.
Screeningul familial este foarte important, având în vedere severitatea bolii și necesitatea instituirii precoce a tratamentului în cursul bolii. Fraţii și surorile unui pacient cu boală Wilson au un risc de 25% de a avea boala, atunci când părinţii sunt sănătoși. Riscul copiilor unui pacient index de a face boala este de aproximativ 0,5%, ţinând cont de frecvenţa generală a purtătorilor de mutaţii. Dacă nu se cunoaște mutaţia cauzatoare de boală la pacientul index, se recomandă efectuarea analizei genetice de haplotip pornind de la părinţi și pacientul index (7).
1. Kilpe VE, Krakauer H, Wren RE. An analysis of liver transplant experience from 37 transplant centers as reported to medicare. Transplantation 1993; 56: 554-61
2. Reilly M, Daly L, Hutchinson M. An epidemiological study of Wilson’s disease in the Republic of Ireland. J Neurol Neurosurg Psychiatry. 1993 Mar;56(3):298-300
3. Steindl P, Ferenci P, Dienes HP et al. Wilson’s disease in patients presenting with liver disease: a diagnostic challenge. Gastroenterology. 1997 Jul;113(1):212-8
4. Ferenci P, Caca K, Loudianos G et al. Diagnosis and phenotypic classification of Wilson disease. Liver Int. 2003 Jun;23(3):139-42
5. Iacob R, Iacob S, Năstase A et al. The His1069Gln mutation in the ATP7B gene în Romanian patients with Wilson’s disease referred to a tertiary gastroenterology center. J Gastrointestin Liver Dis. 2012 Jun;21(2):181-5
6. European Association for Study of Liver. EASL Clinical Practice Guidelines: Wilson’s disease. J Hepatol. 2012 Mar;56(3):671-85
7. Ferenci P. Wilson’s Disease. Clin Gastroenterol Hepatol. 2005 Aug;3(8):726-33
Dacă vrei să fii la curent cu tot ce se întâmplă în lumea medicală, abonează-te la „Viața Medicală”, publicația profesională, socială și culturală a profesioniștilor în Sănătate din România!
Titularii abonamentelor pe 12 luni sunt creditați astfel de:

Dacă vrei să fii la curent cu tot ce se întâmplă în lumea medicală, abonează-te la „Viața Medicală”, publicația profesională, socială și culturală a profesioniștilor în Sănătate din România!
Află mai multe informații despre oferta de abonare.
Cookie-urile ne ajută să vă îmbunătățim experiența pe site-ul nostru. Prin continuarea navigării pe site-ul www.viata-medicala.ro, veți accepta implicit folosirea de cookie-uri pe parcursul vizitei dumneavoastră.
Da, sunt de acord Aflați mai multe