Cele mai frecvente boli cu transmitere genetică ce se manifestă cu alterare structurală cardiacă și aritmii ventriculare sunt cardiomiopatia aritmogenă a ventriculului drept și cardiomiopatia hipertrofică.
Primele manifestări clinice pot fi sincopa, oprirea cardiacă sau chiar moartea subită cardiacă, determinate de prezenţa aritmiilor ventriculare maligne, uneori înainte ca afectările structurale să fie evidente la examinările de rutină.
Cardiomiopatia hipertrofică (CMH) este definită prin creșterea grosimii peretelui ventricular care nu poate fi explicată doar prin condiţii anormale de creștere a sarcinii ventriculului stâng (1). Ea este a doua cauză de deces (moarte subită) la sportivii de performanţă în Italia (2) și prima în Statele Unite ale Americii (3); diferenţele provin probabil din diferenţele programelor de depistare activă.
Hipertrofia ventriculară stângă neexplicată de afecţiuni care cresc sarcina ventriculară are o incidenţă globală de 0,02-0,23%, mai scăzută la persoanele sub 25 de ani (1). În SUA este cea mai frecventă afecţiune cardiacă transmisă genetic, fiind prezentă la una din 500 de persoane.
CMH este o boală cu transmitere cel mai des autozomal dominantă și este determinată în principal de mutaţii ale genelor care determină alterarea proteinelor sarcolemale (cel mai frecvent MYH7, care codifică lanţul greu al betamiozinei, și MYBPC3, care codifică proteina C de legare a miozinei). Mai puţin frecvente sunt mutaţiile genelor care codifică troponinele T și I (TNNI3, TNNT2), lanţul alfa- 1 al tropomiozinei (TPM1) sau lanţul ușor 3 al miozinei (MYL3) (1).
Diagnosticul se pune pe o grosime a peretelui ventricular ≥15 mm, însă pot fi necesare investigaţii suplimentare dacă grosimea peretelui este mai mică, în afecţiuni care se asociază cu CMH.
Mecanismele incriminate în apariţia aritmiilor ventriculare în CMH sunt multiple. Hipertrofia miocitară duce la prelungirea potenţialului de acţiune și la apariţia postpotenţialelor precoce, mecanism dovedit experimental la animale, însă nu și la om. Creșterea frecvenţei cardiace induse de catecolamine și apariţia postpotenţialelor tardive care induc activitate declanșată este un mecanism mai probabil, care poate fi determinat sau accentuat și de ischemia relativă prin creșterea distanţei capilar-fibră musculară. Aceste modificări sunt favorizate de tulburări electrolitice, în special de hipopotasemie. De asemenea, fibroza și dezorganizarea dispunerii miocitelor creează zone de conducere lentă a impulsului electric, care pot întreţine aritmii prin reintrare (4). Experimental, principalul curent ionic implicat în tulburările de repolarizare este ICaL, iar blocarea INaX, ICaL, INaL poate restaura modelul electrofiziologic normal al miocitului (5).
Aritmiile ventriculare sub formă de tahicardii ventriculare nesusţinute apar la un sfert dintre pacienţi și se corelează cu grosimea peretelui ventricular și cu prezenţa de gadoliniu la IRM. Ele sunt un factor de risc pentru moartea subită cardiacă. Aritmiile susţinute se documentează mai rar și trebuie exclusă prezenţa bolii coronariene (6).
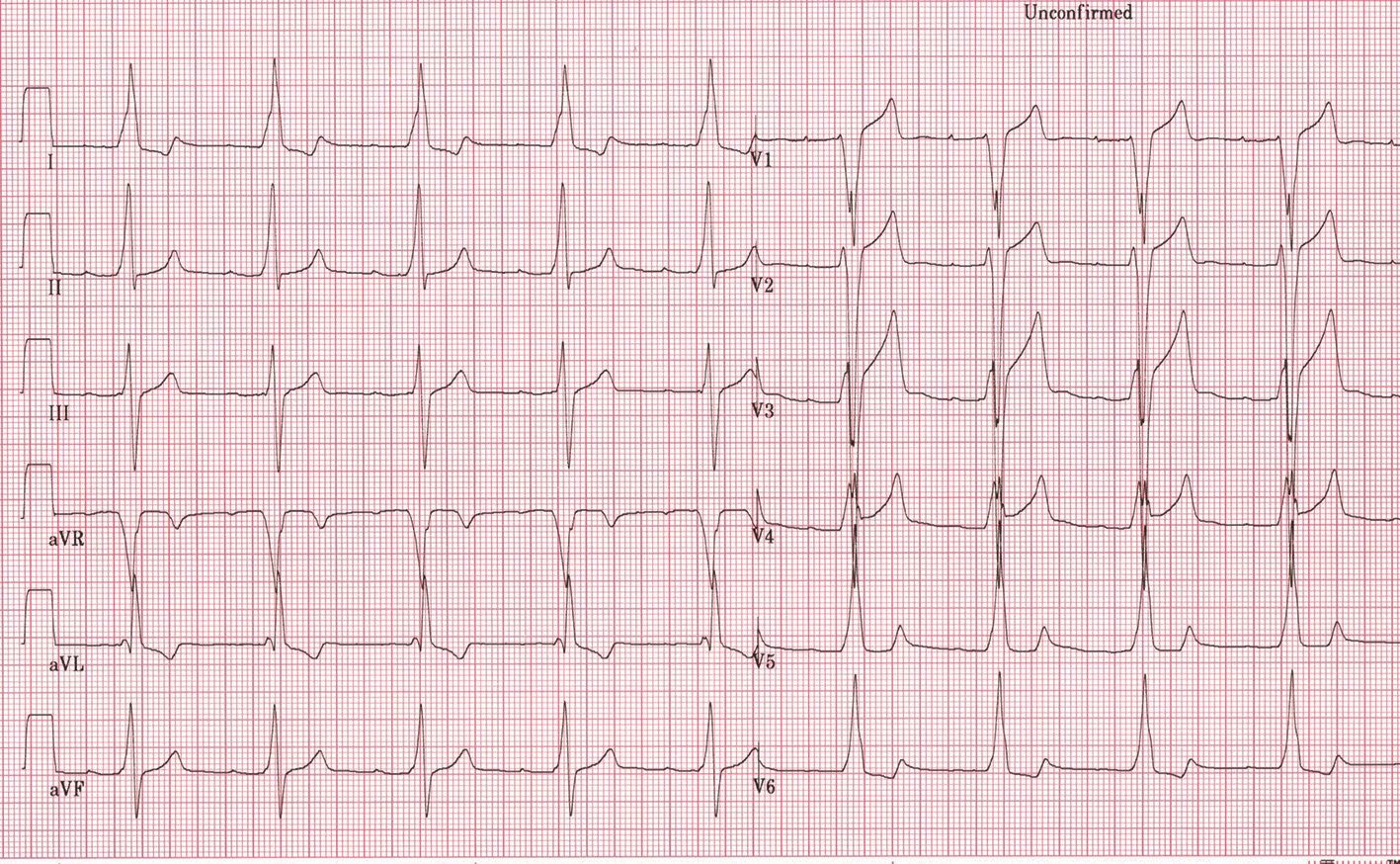
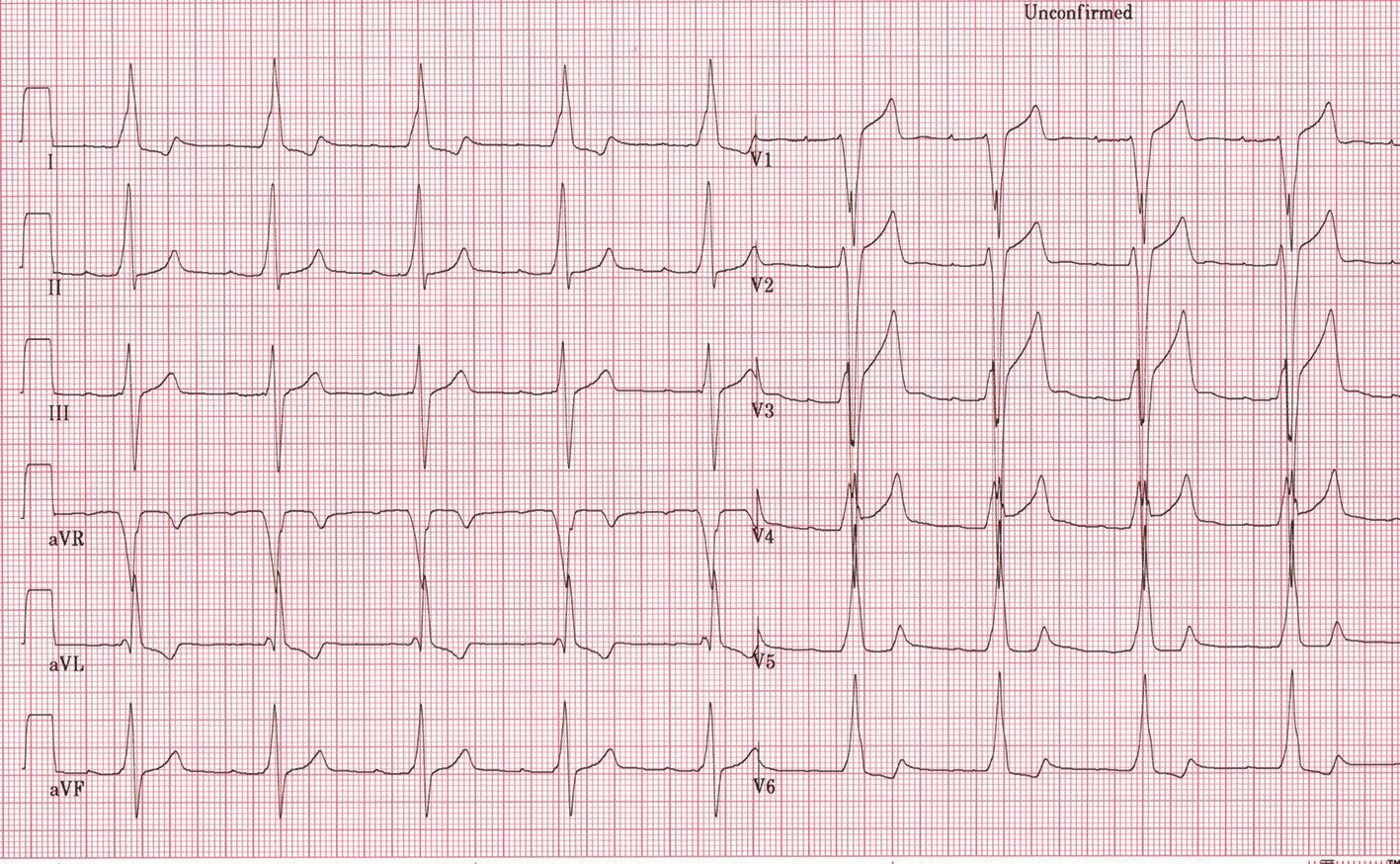
Sincopa în CMH poate fi cauzată de aritmiile ventriculare, indicând necesitatea implantării unui defibrilator cardiac, dar și de aritmiile supraventriculare cu conducere rapidă, de obstrucţia în tractul de ejecţie al ventriculului stâng sau de blocurile atrioventriculare. Tulburările de conducere atrioventriculare progresive apar mai frecvent în tulburările mitocondriale, în anumite tezaurismoze (boala Andersson-Fabry), desminopatii și la pacienţii cu mutaţii PRKAG2 (1). Asocierea cu preexcitaţia ventriculară nu este rară. De asemenea, tulburări ale conducerii impulsului electric pot apărea în tulburări metabolice, așa cum este boala Danon, determinată de afectarea LAMP-2; acestea pot fi sub forma preexcitaţiei ventriculare și a tulburărilor de conducere atrioventriculare (figura).
Conform recomandărilor Societăţii Europene de Cardiologie (6), evaluarea riscului de moarte subită cardiacă se face cu ajutorul calculatorului de risc la cinci ani, existent online http://doc2do.com/hcm/webHCM.html (indicaţie clasa I, nivel de evidenţă B). Sunt luate în considerare vârsta, grosimea peretelui ventricular, dimensiunile atriului stâng, gradientul în tractul de ejecţie, istoricul familial sau prezenţa sincopelor al căror mecanism este necunoscut. Implantarea unui defibrilator trebuie luată în considerare la pacienţii cu risc de peste 6% la cinci ani (clasa IIa) și poate fi benefică la anumiţi pacienţi cu risc 4-6% (IIb) și la cei care au caracteristici clinice la risc de moarte subită (IIb). Această evaluare se face la momentul diagnosticului și apoi se repetă la 1-2 ani (clasa I); defibrilatorul aduce beneficii certe la pacienţii care au avut oprire cardiacă resuscitată sau care au tahicardii ventriculare ce determină sincopă sau alterare hemodinamică (clasa I); tratamentul medicamentos constă în administrarea de betablocante fără efect vasodilatator; de asemenea, amiodarona ar putea avea un efect în reducerea incidenţei morţii subite cardiace. Betablocantele și disopiramida au efect în reducerea obstrucţiei, însă efectul antiaritmic nu este bine dovedit. La fel ca la pacienţii cu cardiomiopatie aritmogenă a ventriculului drept, este contraindicat sportul de performanţă (clasa IC).
Cardiomiopatia dilatativă (CMD) nonischemică are în 20% dintre cazuri transmitere familială, fiind consecinţa unor mutaţii genetice care determină alterarea proteinelor sarcolemale și desmozomale, iar mutaţiile laminei A/C și ale desminei se asociază cu tulburări de conducere (6). Cea mai frecventă mutaţie a proteinelor sarcolemale este mutaţia titinei (TTN), întâlnită la 25% dintre cazurile familiale și la 18% dintre cazurile de CMD idiopatică (7).
Înainte de implantarea defibrilatoarelor cardiace, aproximativ 50% dintre decesele la pacienţii cu insuficienţă cardiacă aveau drept cauză moartea subită cardiacă de natură aritmică. Factorii de risc pentru moartea subită cardiacă sunt: fragmentarea QRS pe ECG, microalternaţa de undă T și captarea tardivă de gadoliniu la IRM (6).
Aritmia specifică CMD nonischemice este cea prin reintrare pe ramuri, care apare în special la pacienţii cu bloc de ramură stângă și PR lung (la studiul electrofiziologic, alungirea intervalului HV). Pacienţii cu CMD au și alte tahicardii ventriculare: monomorfe, polimorfe și/sau fibrilaţie ventriculară.
Prevenţia morţii subite cardiace prin implantarea unui defibrilator este indicată pentru profilaxia secundară sau la cei cu fracţii de ejecţie ≤ 35% sub tratament optim minimum trei luni și cu o speranţă de viaţă mai mare de un an (6). Deși ghidurile europene menţin în continuare această indicaţie, studiul DANISH a arătat că este posibil să nu existe un beneficiu suficient comparativ cu riscurile pentru a justifica această terapie doar pe baza fracţiei de ejecţie scăzute la cei cu CMD nonischemică (8). Tahicardia prin reintrare pe ramuri este curabilă prin ablaţia pe cateter (clasa I dacă este refractară la terapia medicamentoasă), ce are potenţial și pentru alte aritmii ventriculare (clasă IIb) (6).
1. Elliott PM et al. 2014 ESC Guidelines on diagnosis and management of hypertrophic cardiomyopathy: the Task Force for the Diagnosis and Management of Hypertrophic Cardiomyopathy of the European Society of Cardiology (ESC). Eur Heart J. 2014 Oct 14;35(39):2733-79
2. Corrado D et al. Does sports activity enhance the risk of sudden death in adolescents and young adults? J Am Coll Cardiol. 2003 Dec 3;42(11):1959-63
3. Maron BJ et al. Sudden deaths in young competitive athletes: analysis of 1866 deaths in the United States, 1980-2006. Circulation. 2009 Mar 3;119(8):1085-92
4. Janse MJ, De Bakker JMT. Arrhythmia substrate and management in hypertrophic cardiomyopathy: from molecules to implantable cardioverter–defibrillators. Eur Heart J. 2001;Supl 3:L15-20
5. Passini E et al. Mechanisms of pro-arrhythmic abnormalities in ventricular repolarisation and anti-arrhythmic therapies in human hypertrophic cardiomyopathy. J Mol Cell Cardiol. 2016 Jul;96:72-81
6. Priori SG et al. 2015 ESC Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: The Task Force for the Management of Patients with Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death of the European Society of Cardiology (ESC). Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC). Eur Heart J. 2015 Nov 1;36(41):2793-2867
7. Mestroni L et al. Genetic causes of dilated cardiomyopathy. Prog Pediatr Cardiol. 2014 Dec;37(1-2):13-18
8. Kober L et al. Defibrillator Implantation in Patients with Nonischemic Systolic Heart Failure. N Engl J Med. 2016 Sep 29;375(13):1221-30
Dacă vrei să fii la curent cu tot ce se întâmplă în lumea medicală, abonează-te la „Viața Medicală”, publicația profesională, socială și culturală a profesioniștilor în Sănătate din România!
Titularii abonamentelor pe 12 luni sunt creditați astfel de:

Dacă vrei să fii la curent cu tot ce se întâmplă în lumea medicală, abonează-te la „Viața Medicală”, publicația profesională, socială și culturală a profesioniștilor în Sănătate din România!
Află mai multe informații despre oferta de abonare.
Cookie-urile ne ajută să vă îmbunătățim experiența pe site-ul nostru. Prin continuarea navigării pe site-ul www.viata-medicala.ro, veți accepta implicit folosirea de cookie-uri pe parcursul vizitei dumneavoastră.
Da, sunt de acord Aflați mai multe