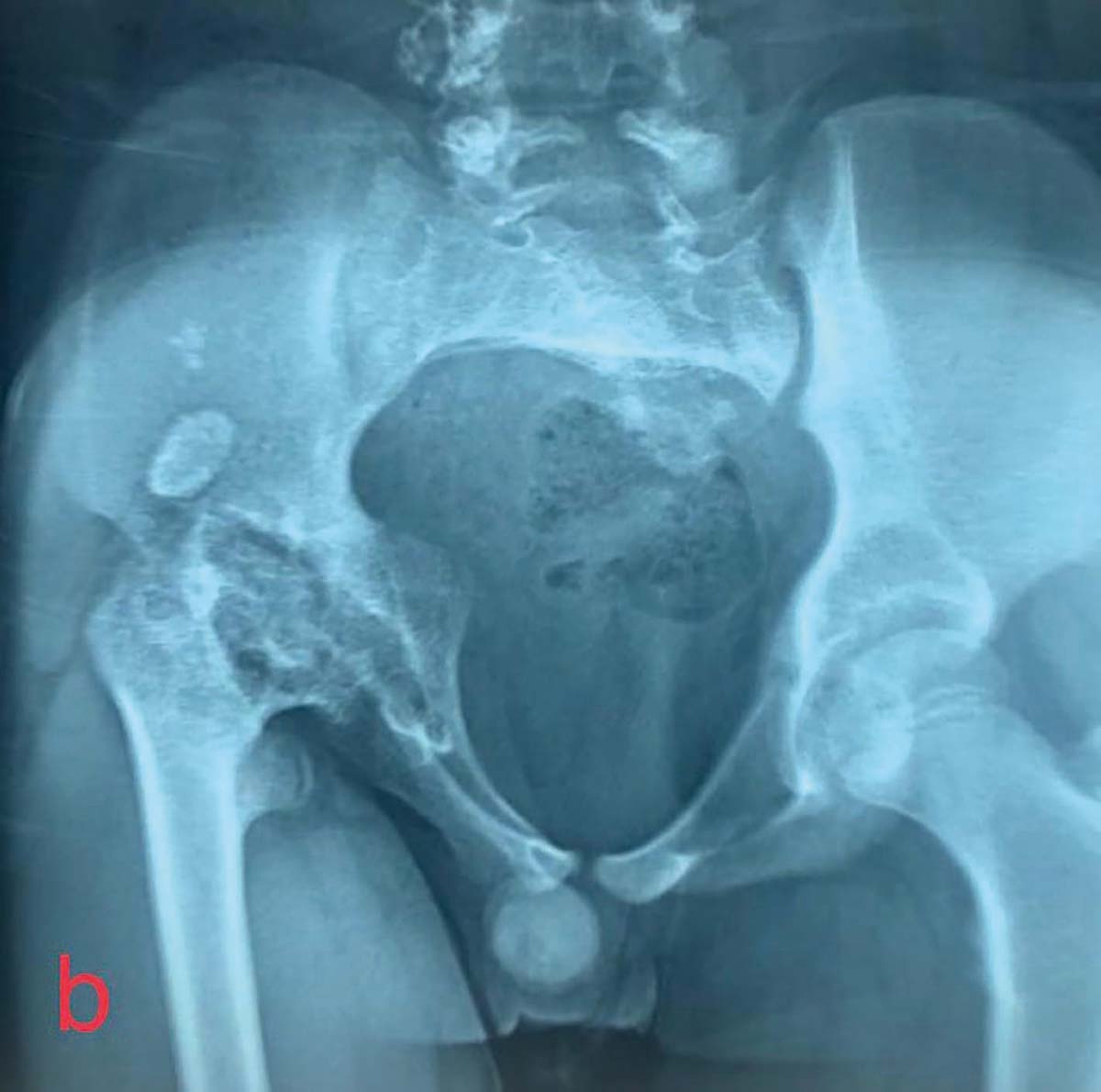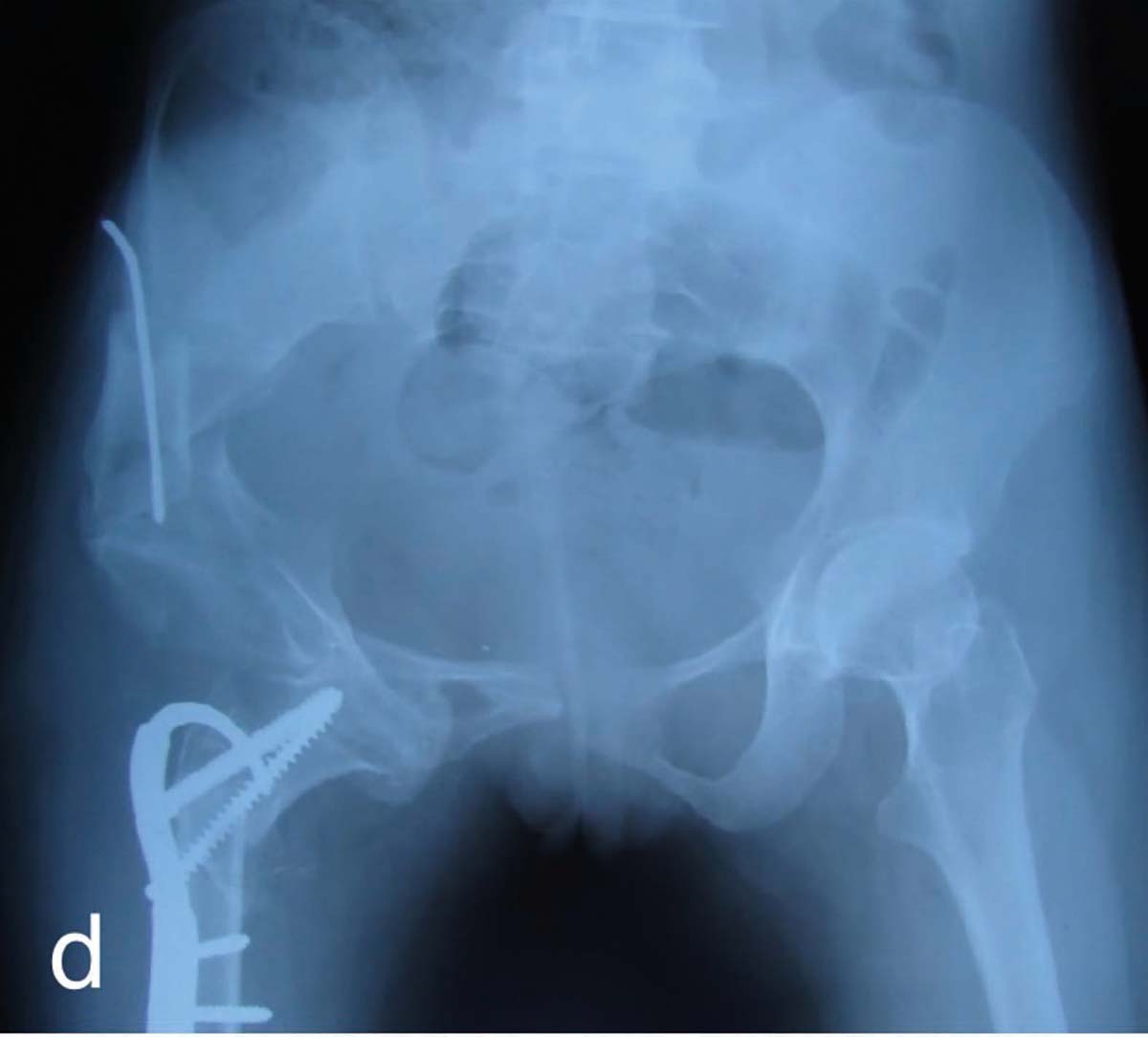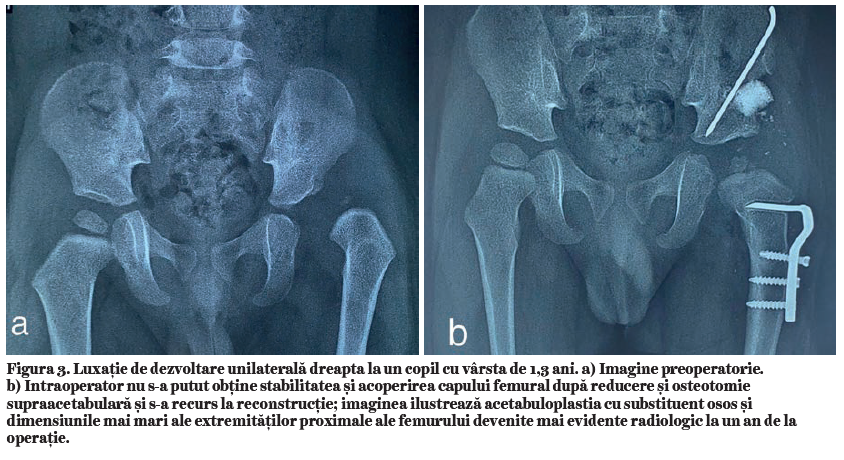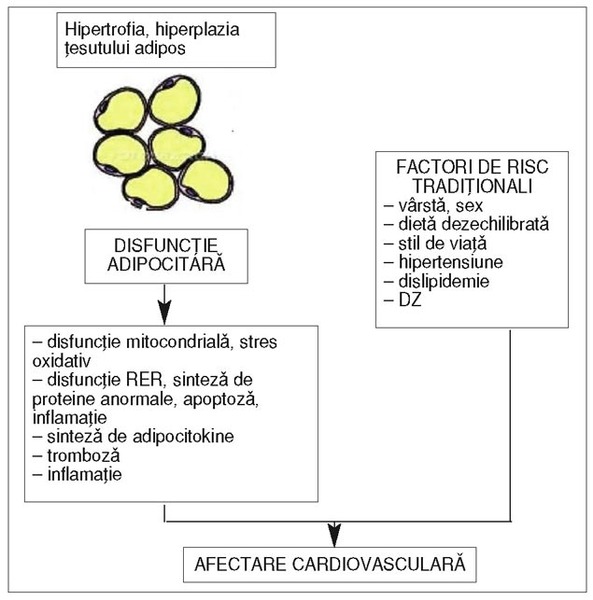
 Disfuncţia adipocitară. Pe scurt, disfuncţia
adipocitară poate fi descrisă astfel (3): dezechilibrul energetic (aport
excesiv de nutrienţi) determină creşterea nivelurilor serice de glucoză,
trigliceride şi implicit de acizi graşi liberi, care conduc la hipertrofia şi
hiperplazia adipocitelor; capacitatea ţesutului adipos de a prelua aceşti
nutrienţi este depăşită şi se declanşează o cascadă de evenimente inflamatorii (infiltrarea ţesutului
adipos cu monocito-macrofage, creşterea secreţiei de citokine proinflamatorii)
şi secretorii (creşterea secreţiei de
adipokine), care conduc în principal la insulinorezistenţă prin inactivarea
receptorului insulinic pe calea c-Jun N-terminal kinazei (JNK) – aceasta
fosforilează substratul receptorului de insulină 1 (IRS1) la nivelul serinei
312, determinând inactivarea transmiterii intracelulare a semnalului
insulinic. În plus, în mod direct, atât acizii graşi liberi, cât şi o serie de
adipokine (ex. AFABP4) determină suprasolicitarea reticulului endoplasmic,
ceea ce conduce la: disfuncţia sa, acumularea de proteine incorect conformate,
apoptoza adipocitelor şi implicit la amplificarea cascadei inflamatorii (fig. 2). Disfuncţia reticulului
endoplasmic, precum şi acumularea acizilor graşi liberi determină suplimentar
stres oxidativ mitocondrial, cu producerea speciilor reactive de oxigen, care
accentuează insulinorezistenţa prin scăderea transportului intracelular al
glucozei (4); se declanşează astfel un cerc vicios, deoarece glucoza în exces
determină la rândul său accentuarea producerii de radicali liberi.
Disfuncţia adipocitară. Pe scurt, disfuncţia
adipocitară poate fi descrisă astfel (3): dezechilibrul energetic (aport
excesiv de nutrienţi) determină creşterea nivelurilor serice de glucoză,
trigliceride şi implicit de acizi graşi liberi, care conduc la hipertrofia şi
hiperplazia adipocitelor; capacitatea ţesutului adipos de a prelua aceşti
nutrienţi este depăşită şi se declanşează o cascadă de evenimente inflamatorii (infiltrarea ţesutului
adipos cu monocito-macrofage, creşterea secreţiei de citokine proinflamatorii)
şi secretorii (creşterea secreţiei de
adipokine), care conduc în principal la insulinorezistenţă prin inactivarea
receptorului insulinic pe calea c-Jun N-terminal kinazei (JNK) – aceasta
fosforilează substratul receptorului de insulină 1 (IRS1) la nivelul serinei
312, determinând inactivarea transmiterii intracelulare a semnalului
insulinic. În plus, în mod direct, atât acizii graşi liberi, cât şi o serie de
adipokine (ex. AFABP4) determină suprasolicitarea reticulului endoplasmic,
ceea ce conduce la: disfuncţia sa, acumularea de proteine incorect conformate,
apoptoza adipocitelor şi implicit la amplificarea cascadei inflamatorii (fig. 2). Disfuncţia reticulului
endoplasmic, precum şi acumularea acizilor graşi liberi determină suplimentar
stres oxidativ mitocondrial, cu producerea speciilor reactive de oxigen, care
accentuează insulinorezistenţa prin scăderea transportului intracelular al
glucozei (4); se declanşează astfel un cerc vicios, deoarece glucoza în exces
determină la rândul său accentuarea producerii de radicali liberi. În ceea ce priveşte
instituirea complicaţiilor cardiovasculare ale obezităţii, modificarea
princeps o constituie apariţia disfuncţiei endoteliale. La baza acesteia stau două mecanisme etiopatogenice majore – hiperinsulinemia (consecinţa rezistenţei
la insulină) şi statusul proinflamator
cronic (fig. 3). În fiecare din
aceste mecanisme, adipocitokinele secretate la nivelul ţesutului adipos au un
rol etiopatogenic (direct sau indirect).
În ceea ce priveşte
instituirea complicaţiilor cardiovasculare ale obezităţii, modificarea
princeps o constituie apariţia disfuncţiei endoteliale. La baza acesteia stau două mecanisme etiopatogenice majore – hiperinsulinemia (consecinţa rezistenţei
la insulină) şi statusul proinflamator
cronic (fig. 3). În fiecare din
aceste mecanisme, adipocitokinele secretate la nivelul ţesutului adipos au un
rol etiopatogenic (direct sau indirect). adeziune a celulelor vasculare), cu efect
proaderent; • în plus, insulina determină producerea de specii reactive de
oxigen, mecanismul molecular al acestui efect nefiind încă complet cunoscut
(5). În concentraţii fiziologice, speciile reactive de oxigen conduc la
creşterea producerii de NO, favorizând semnalizarea insulin-IR2-PI3 kinaza;
în concentraţii excesive însă, speciile reactive de oxigen determină insulinorezistenţă
şi disfuncţie endotelială (5). Mecanismul-cheie ce explică legătura dintre
hiperinsulinismul din obezitate şi disfuncţia endotelială este rezistenţa selectivă la insulină, care
se adresează doar căii de semnalizare PI3K, în timp ce căile dependente de MAPK
nu sunt afectate (6–8).
adeziune a celulelor vasculare), cu efect
proaderent; • în plus, insulina determină producerea de specii reactive de
oxigen, mecanismul molecular al acestui efect nefiind încă complet cunoscut
(5). În concentraţii fiziologice, speciile reactive de oxigen conduc la
creşterea producerii de NO, favorizând semnalizarea insulin-IR2-PI3 kinaza;
în concentraţii excesive însă, speciile reactive de oxigen determină insulinorezistenţă
şi disfuncţie endotelială (5). Mecanismul-cheie ce explică legătura dintre
hiperinsulinismul din obezitate şi disfuncţia endotelială este rezistenţa selectivă la insulină, care
se adresează doar căii de semnalizare PI3K, în timp ce căile dependente de MAPK
nu sunt afectate (6–8). celule adipoase creşterea eliberării de adipocitokine
inflamatorii (13).
celule adipoase creşterea eliberării de adipocitokine
inflamatorii (13). insulină cu toate consecinţele sale metabolice şi cardiovasculare), dar şi a
stresului oxidativ şi a reacţiei inflamatorii locale şi sistemice ce
caracterizează obezitatea. Toate aceste fenomene conduc la ateromatoză, dar şi
la instabilitatea plăcilor de aterom cu tromboză, deci la eveniment cardiovascular
major. Cu alte cuvine, mediatorii sintetizaţi la nivelul ţesutului adipos în
ansamblul său (celulele adipoase în diversele lor stadii de maturare şi
componentele celulare stromale şi inflamatorii – celulele endoteliale,
fibroblastele, sistemul monocito-macrofagic) sunt nu doar iniţiatorii, ci şi
actorii finali în filmul evenimentelor care conduc la deznodământul
cardiovascular (fig. 6).
insulină cu toate consecinţele sale metabolice şi cardiovasculare), dar şi a
stresului oxidativ şi a reacţiei inflamatorii locale şi sistemice ce
caracterizează obezitatea. Toate aceste fenomene conduc la ateromatoză, dar şi
la instabilitatea plăcilor de aterom cu tromboză, deci la eveniment cardiovascular
major. Cu alte cuvine, mediatorii sintetizaţi la nivelul ţesutului adipos în
ansamblul său (celulele adipoase în diversele lor stadii de maturare şi
componentele celulare stromale şi inflamatorii – celulele endoteliale,
fibroblastele, sistemul monocito-macrofagic) sunt nu doar iniţiatorii, ci şi
actorii finali în filmul evenimentelor care conduc la deznodământul
cardiovascular (fig. 6). este singura adipokină cu rol protector
metabolic şi cardiovascular, precum şi singura a cărei sinteză este scăzută în
obezitate.
este singura adipokină cu rol protector
metabolic şi cardiovascular, precum şi singura a cărei sinteză este scăzută în
obezitate.
Bibliografie
1. http://epp.eurostat.ec.europa.eu/statistics_explained/index.php/Overweight_and_obesity_-_BMI_statistics
2. http://www.easo.org/childhood-obesity-cotf/facts-a-statistics. 2013
3. Balagopal PB, de Ferranti SD, Cook S, Daniels SR, Gidding SS, Hayman LL, McCrindle BW, Mietus-Snyder ML, Steinberger J; American Heart Association Committee on Atherosclerosis Hypertension and Obesity in Youth of the Council on Cardiovascular Disease in the Young; Council on Nutrition, Physical Activity and Metabolism; Council on Epidemiology and Prevention. Nontraditional risk factors and biomarkers for cardiovascular disease: mechanistic, research, and clinical considerations for youth: a scientific statement from the American Heart Association. Circulation. 2011 Jun 14;123(23):2749-69
4. de Ferranti S, Mozaffarian D. The perfect storm: obesity, adipocyte dysfunction, and metabolic consequences. Clin Chem. 2008 Jun;54(6):945-55.
5. Bastard JP, Maachi M, Lagathu C, Kim MJ, Caron M, Vidal H, Capeau J, Feve B. Recent advances in the relationship between obesity, inflammation, and insulin resistance. Eur Cytokine Netw 2006 Mar;17(1):4-12.
6. Cusi K, Maezono K, Osman A, Pendergrass M, Patti ME, Pratipanawatr T, DeFronzo RA, Kahn CR, Mandarino LJ. Insulin resistance differentially affects the PI 3-kinase- and MAP kinase-mediated signaling in human muscle. J Clin Invest. 2000 Feb;105(3):311-20.
7. Itani SI, Ruderman NB, Schmieder F, Boden G. Lipid-induced insulin resistance in human muscle is associated with changes in diacylglycerol, protein kinase C, and IkappaB-alpha. Diabetes. 2002 Jul;51(7):2005-11.
8. Montagnani M, Golovchenko I, Kim I, Koh GY, Goalstone ML, Mundhekar AN, Johansen M, Kucik DF, Quon MJ, Draznin B. Inhibition of phosphatidylinositol 3-kinase enhances mitogenic actions of insulin in endothelial cells. J Biol Chem. 2002 Jan 18;277(3):1794-9.
9. Hotamisligil GS. Inflammation and metabolic disorders. Nature 2006 Dec 14;444(7121):860-7.
10. Trayhurn P, Wang B, Wood IS. Hypoxia in adipose tissue: a basis for the dysregulation of tissue function in obesity? Br J Nutr. 2008 Aug;100(2):227-35.
11. Yin J, Gao Z, He Q, Zhou D, Guo Z, Ye J. Role of hypoxia in obesity-induced disorders of glucose and lipid metabolism in adipose tissue. Am J Physiol Endocrinol Metab. 2009 Feb;296(2):E333-42.
12. Tsukumo DM, Carvalho-Filho MA, Carvalheira JB, Prada PO, Hirabara SM, Schenka AA, Ara˙jo EP, Vassallo J, Curi R, Velloso LA, Saad MJ. Loss-of-function mutation in Toll-like receptor 4 prevents diet-induced obesity and insulin resistance. Diabetes. 2007 Aug;56(8):1986-98.
13. Lee JY, Sohn KH, Rhee SH, Hwang D. Saturated fatty acids, but not unsaturated fatty acids, induce the expression of cyclooxygenase-2 mediated through Toll-like receptor 4. J Biol Chem. 2001 May 18;276(20):16683-9.
14. Schaeffler A, Gross P, Buettner R, Bollheimer C, Buechler C, Neumeier M, Kopp A, Schoelmerich J, Falk W. Fatty acid-induced induction of Toll-like receptor-4/nuclear factor-kappaB pathway in adipocytes links nutritional signalling with innate immunity. Immunology. 2009 Feb;126(2):233-45
15. Steer P, Basu S, Lithell H, Vessby B, Berne C, Lind L. Acute elevations of medium- and long-chain fatty acid have different impacts on endothelium-dependent vasodilation in humans. Lipids. 2003 Jan;38(1):15-9.
16. Sarafidis PA, Bakris GL. Insulin resistance, hyperinsulinemia, and hypertension: an epidemiologic approach. J Cardiometab Syndr. 2006 Fall;1(5):334-42.
17. Esenabhalu VE, Schaeffer G, Graier WF. Free fatty acid overload attenuates Ca2+ signaling and NO production in endothelial cells. Antioxid Redox Signal. 2003 Apr;5(2):147-53.
18. Li H, Li H, Bao Y, Zhang X, Yu Y. Free fatty acids induce endothelial dysfunction and activate protein kinase C and nuclear factor-kB pathway in rat aorta. Int J Cardiol. 2011 Oct 20;152(2):218-24.
19. Stepniakowski KT, Lu G, Davda RK, Egan BM. Fatty acids augment endothelium-dependent dilation in hand veins by a cyclooxygenase-dependent mechanism. Hypertension. 1997 Dec;30(6):1634-9.
20. Lopes HF, Martin KL, Nashar K, Morrow JD, Goodfriend TL, Egan BM. DASH diet lowers blood pressure and lipid-induced oxidative stress in obesity. Hypertension. 2003 Mar;41(3):422-30.
21. Artwohl M, Roden M, Waldhausl W, Freudenthaler A, Baumgartner-Parzer SM. Free fatty acids trigger apoptosis and inhibit cell cycle progression in human vascular endothelial cells. FASEB J. 2004 Jan;18(1):146-8.
22. Di Chiara T, Argano C, Corrao S, Scaglione R, Licata G. Hypoadiponectinemia: A Link between Visceral Obesity and Metabolic Syndrome. J Nutr Metab. 2012;2012:175245.
Dacă vrei să fii la curent cu tot ce se întâmplă în lumea medicală, abonează-te la „Viața Medicală”, publicația profesională, socială și culturală a profesioniștilor în Sănătate din România!
Titularii abonamentelor pe 12 luni sunt creditați astfel de:

Dacă vrei să fii la curent cu tot ce se întâmplă în lumea medicală, abonează-te la „Viața Medicală”, publicația profesională, socială și culturală a profesioniștilor în Sănătate din România!
Află mai multe informații despre oferta de abonare.
Cookie-urile ne ajută să vă îmbunătățim experiența pe site-ul nostru. Prin continuarea navigării pe site-ul www.viata-medicala.ro, veți accepta implicit folosirea de cookie-uri pe parcursul vizitei dumneavoastră.
Da, sunt de acord Aflați mai multe